氣相色譜法分析丙烯腈反應器流出物中丙烯腈、乙腈、丙烯醛、丙烯酸
2019-06-03 07:54:56吳澤學
安徽化工 2019年2期
關鍵詞:分析
吳澤學
(中國石油化工股份有限公司安慶分公司檢驗計量中心,安徽安慶246002)
工業用丙烯腈主要用于生產腈綸纖維、ABS 塑料和SAN 樹脂[1]。中石化安慶分公司丙烯腈裝置采用Sohio公司丙烯氨氧化先進工藝技術[2-4],國內采用此技術的丙烯腈生產裝置共有13 套。丙烯腈反應器流出物的組成分析是計算丙烯腈收率、丙烯轉化率,評價催化劑性能的重要依據。丙烯腈反應器流出物的組成包括丙烯腈、乙腈、丙烯醛、丙烯酸、吡啶、反丁烯二腈、丙烯酰胺、丁二腈、一氧化碳、二氧化碳、丙烯、丙烷、氧氣等有機和無機雜質[5]。現有丙烯腈反應器流出物中丙烯腈、乙腈、丙烯醛、丙烯酸的分析方法為使用兩臺色譜儀,一臺裝有PQ(80~100 目)2 m 長不銹鋼柱用于分析丙烯腈、乙腈、丙烯醛,另一臺裝有PQS(80~100 目)1 m 長玻璃柱用于分析丙烯酸。國內也有使用Carbowax 20 M涂裝4%Carbopack D-BA 的玻璃填充柱,采用程序升溫來分析的方法[6],這類方法的缺點是使用一段時間后保留值改變,分離度下降且分析時間較長;乙腈與丙烯醛分離效果不好;而且需要兩次進樣及外標法定量,然而進樣量及色譜條件如溫度、流量的變化,容易影響樣品分析結果的準確度。本方法為采用一臺氣相色譜,選用DB-FFAP(60 m×0.32 mm×0.50 μm)毛細管色譜柱,柱流量3.0 mL/min,分流比50∶1,采用程序升溫及內標法定量,實現了一次進樣同時分析丙烯腈反應器流出物中丙烯腈、乙腈、丙烯醛、丙烯酸,克服了現有分析方法需要兩臺氣相色譜才能分別檢測流出物中丙烯腈、乙腈、丙烯醛、丙烯酸的不足。本方法不僅縮短分析時間至20 min,而且明顯改善了乙腈與丙烯醛的分離效果,減少了丙烯酸的吸附,各組分相對誤差在-1.55%~+0.88%之間,滿足了生產實際要求。
1 實驗部分
1.1 材料與試劑
島津GC-2014 氣相色譜(具SPL 分流裝置、氫火焰檢測器);美國Thermo Atlas 8.1 色譜工作站;梅特勒托利多ME-204 電子天平;10 μL 注射器;50 mL 容量瓶。
丙烯腈(99.0%,國藥集團化學試劑有限公司);丙烯醛(90%,SIGMA-ALDRICH);2- 丁酮(99.5%,國藥集團化學試劑有限公司);乙腈(99.8%,國藥集團化學試劑有限公司);丙烯酸(99%,東京化成工業株式會社);甲酸(99.5%,國藥集團化學試劑有限公司)。
1.2 標樣的配制
取一潔凈、干燥恒重的50 mL 容量瓶,根據樣品實際濃度配制濃度近似的反應器流出物標樣,依次加入約50 mL 二級蒸餾水、240 μL 色譜純丙烯腈、10 μL 色譜純乙腈、7 μL 色譜純丙烯酸、5 μL 色譜純丙烯醛、40 μL 色譜純2- 丁酮,2- 丁酮作為內標物[7],并分別在電子天平上準確稱量,混勻,在室溫下放置10 min。標樣配制結果見表1。
1.3 色譜條件
色譜柱DB-FFAP(60 m×0.32 mm×0.50 μm)毛細管色譜柱,柱流量3.0 mL/min,分流比50∶1,量程×1,柱初始溫度54℃,保持時間8 min,升溫速率30℃/min,終溫200℃,保持時間8 min,進樣器溫度230℃,檢測器(FID)溫度250℃,氫氣壓力60 kPa,空氣壓力50 kPa,MAKE UP 壓力75 kPa。
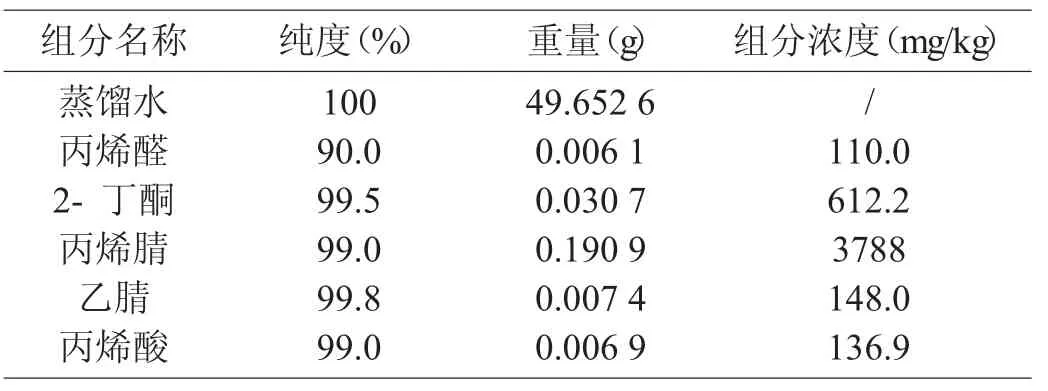
表1 丙烯腈反應器流出物標樣配制明細表
1.4 分析步驟及定量方法
對現配制的丙烯腈反應器流出物標樣進樣,每次2 μL,共進五次標樣,采用內標法定量[8],根據公式計算出丙烯醛、丙烯腈、乙腈、丙烯酸的相對質量校正因子,求出其平均值,結果見表2。
由于丙烯腈具有不穩定的雙鍵結構,極易發生自聚、共聚、水解等[9],丙烯醛、丙烯酸極不穩定,可發生聚合反應,丙烯酸在FFAP 柱上有輕微吸附,故在實際分析過程中,應嚴格控制采樣量及樣品吸收溫度,采樣后應及時分析,分析前進2 至3 針丙烯腈反應器流出物樣品,以減少毛細管柱對丙烯酸的吸附。

表2 各組分的相對質量校正因子
2 結果與討論
2.1 色譜條件的優化
在色譜操作條件中,柱溫、載氣流速、分流比、進樣量的選擇對色譜組分的分離及分析時間有很大影響,色譜柱的選擇與待測組分的分離和保留時間等密切相關,定量方法的選擇則直接影響樣品分析的準確度。
2.1.1 色譜柱選擇
乙腈、丙烯酸的極性強,丙烯酸的沸點高,采用色譜法分離時,色譜柱選擇不恰當,會導致色譜峰保留時間過長,色譜峰拖尾及峰型不對稱等現象。通過對弱極性色譜柱HP-5(30 m×0.32 mm×0.25 μm)和中等極性色譜柱DB-FFAP(60 m×0.32 mm×0.50 μm)的實驗比較,HP-5 色譜柱柱分析時間短,丙烯酸組分峰對稱、拖尾小,但分離效果差,相比較而言,DB-FFAP 色譜柱操作條件范圍廣,分離效果好,但丙烯酸有輕微拖尾現象,故選擇DB-FFAP 色譜柱較恰當。
通過實驗反復比較,本方法色譜條件(見1.3)對丙烯腈反應器流出物丙烯腈、乙腈、丙烯醛、丙烯酸的分離有較好的效果,采用內標法定量,分析結果準確度高。
2.1.2 色譜柱老化
使用前將毛細管柱的頭尾各裁掉5~10 cm,進樣口端與色譜儀的進樣口相連,毛細管柱另一端不與檢測器連接,通入載氣,設置載氣柱流量為1.0 mL/min,依次將柱箱溫度設置為100℃、180℃、220℃各老化1 h。分段老化結束后,將色譜參數設置到正常操作條件(見1.3),將毛細管柱另一端與檢測器連接,注入7 針2 μL 現配的0.1%甲酸溶液,每次進樣間隔10 min,這樣做一方面減少丙烯酸色譜峰拖尾,另一方面使毛細管柱保持在酸性環境中,以減少毛細管柱對丙烯酸的輕微吸附作用。
2.1.3 定量方法優化
本方法采用內標法代替了外標法定量,克服了色譜條件中諸如柱溫、載氣流速、進樣量等的變化對樣品分析結果準確度的影響。
2.2 與現有方法比較
丙烯腈反應器流出物標樣現有分析方法色譜圖分別見圖1、圖2,采用DB-FFAP 毛細管柱,分析丙烯腈反應器流出物標樣色譜圖見圖3。由圖1、圖2、圖3 可知,本實驗方法較現有分析方法節約了分析時間,由45 min減少到20 min,峰型更對稱,減小了丙烯腈、丙烯酸的峰型擴展,改善了乙腈與丙烯醛的分離效果。由于采用一次進樣代替原來的兩次進樣,內標法代替外標法定量,減小了樣品的分析誤差。
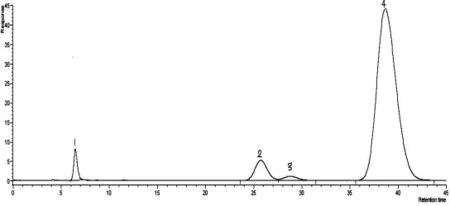
圖1 PQ(80~100 目)填充柱分析丙烯腈反應器流出物標樣色譜圖
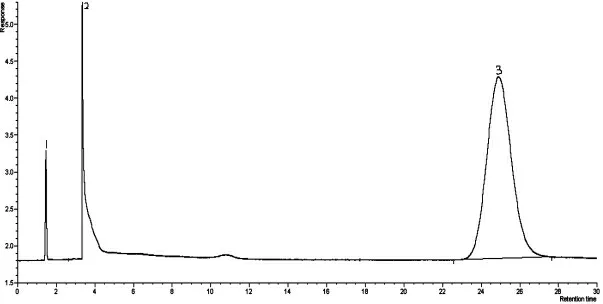
圖2 PQS(80~100 目)玻璃柱分析丙烯腈反應器流出物標樣色譜圖
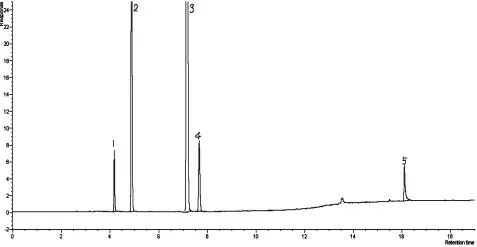
圖3 DB-FFAP 毛細管柱分析丙烯腈反應器流出物標樣色譜圖
2.3 準確度與精密度試驗
對現配制的丙烯腈流出物標樣進樣,每次2 μL,共進五次標樣,采用內標法定量,計算出標樣各組分的相對誤差和相對標準偏差RSD[10],結果見表4。由表4 可知,采用毛細管色譜法,樣品各組分相對誤差為-1.55%~+0.88%,回收率為98.5%~100.9%,相對誤差均小于5%,準確度良好。從測定結果看,樣品相對標準偏差均小于2%。本方法有較好的重復性,可滿足實際分析要求。
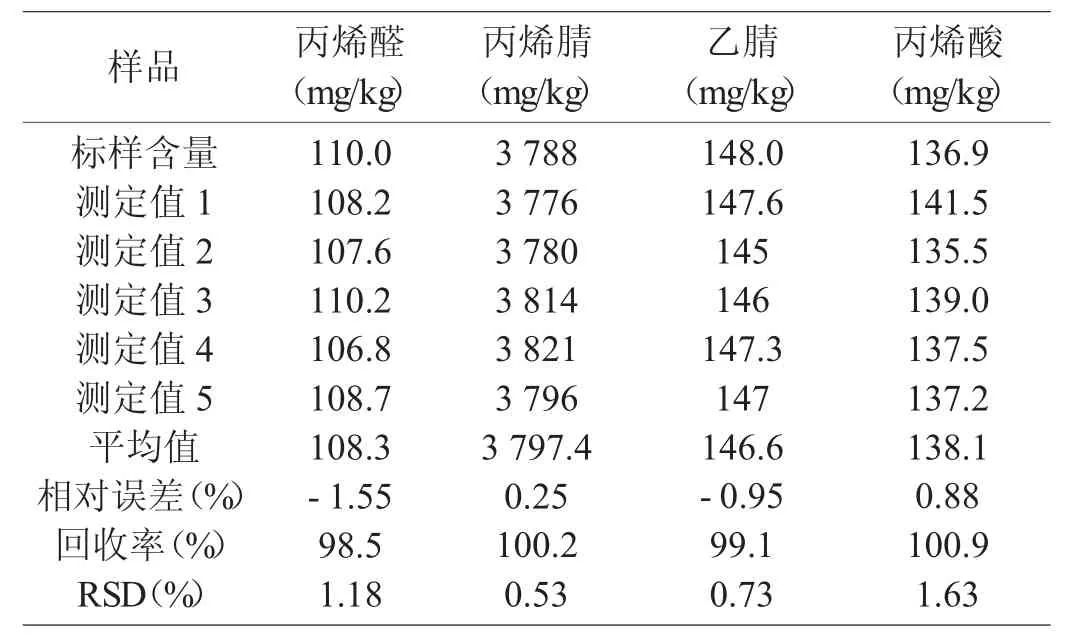
表4 標準樣品準確度和精密度的驗證
2.4 實際樣品分析

圖4 DB-FFAP 毛細管柱分析丙烯腈反應器流出物實際樣品色譜圖
采用上述方法,用采集丙烯腈反應器吸收后的實際流出物樣品,通過色譜分析,發現丙烯腈、乙腈、丙烯醛、丙烯酸分離效果較好,樣品分析的重復性良好,樣品典型色譜圖見圖4。
3 結論
(1)本方法改進了現有的丙烯腈反應器流出物測定方法,使用一臺色譜代替兩臺色譜,毛細管色譜柱代替填充柱,操作簡單,易掌握。
(2)采用DB-FFAP(60 m×0.32 mm×0.50 μm)毛細管色譜柱,通過程序升溫實現一次分析成功。由于毛細管色譜柱分離效率高,縮短分析時間至20 min,提高了樣品組分乙腈與丙烯醛的分離度,減少了丙烯酸的吸附。
(3)采用內標法定量,由兩次進樣改為一次,相對誤差為-1.55%~+0.88%,回收率為98.5%~100.9%,準確度良好,相對標準偏差均小于2%,樣品重復性好,使得丙烯腈單收數據準確可靠,為裝置平穩運行和催化劑評價提供重要依據。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
財經界(學術版)(2015年20期)2015-12-23 09:20:13
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
