Wiedemann-steiner綜合征伴發育遲緩KMT2A基因變異分析一例
2019-04-16 12:03:20曹旭英崔岳崇
實用婦科內分泌雜志(電子版) 2019年35期
關鍵詞:檢測
曹旭英,黃 軻,崔岳崇
(1.義烏市婦幼保健院兒童保健部,浙江 義烏 322000;2.浙江大學醫學院附屬兒童醫院內分泌科,浙江 杭州 310003)
Wiedemann-steiner綜合征是一種罕見的常染色體顯性遺傳病,自韋德曼等人[1]首次報告以來(OMIM 605130),迄今所報告的WDSTS病例,僅描述了三十幾例患者。除了矮小、面部畸形、精神運動延遲、智力低下等非特異性表征外,WDSTS還表現為多毛癥。最近,3項研究發現MLL/KMT2A基因突變導致組胺甲基轉移酶活性不足是WDSTS的原因。MLL/KMT2A基因的基因改變范圍很廣,包括錯義突變、移碼插入或缺失、無義突變和外顯子模型[2-4]。但目前關于其確切遺傳機制仍不清楚。
在本研究中,我們描述了一位女性患兒的臨床表現,并應用高精度臨床外顯PLUS的檢測對其進行基因變異分析,最終確認其為Wiedemann-steiner綜合征KMT2A基因雜合變異。
1 對象與方法
1.1 對象
患兒,女,6歲6個月,生后即出現身高體重增加緩慢,于2018年8月于義烏市婦幼保健院兒童內分泌專科就診。患兒系第1胎第1產,足月順產,出生體重2.1kg,否認圍產期缺氧窒息。母妊娠期正常,無毒物接觸史,父母體健,非近親結婚,父親身高175cm,母親身高158cm,有1弟(4周5月齡,身高102cm,智力發育正常),家族中無遺傳性疾病患者。體格檢查:身高98.5 cm(小于-3SD),體重12.5kg,頭圍46cm,體型消瘦。特殊面容:拱形眉,小下頜,高顎弓,牙齒稀疏22顆(圖1)。心肺聽診未見明顯異常,肝脾肋下未及。輔助檢查:染色體46XX,未見明顯異常。神經發育史:母親回憶患兒4月會抬頭,12個月扶走,15個月能獨走,語言發育落后,24個月會3-4個單詞,現詞匯量少,理解能力較差,膽小,不合群。測試韋氏量表得分59,SM得分9。曾行生長激素激發試驗結果正常,給予喂養指導,依舊改善不明顯。
1.2 方法:
1.2.1 基礎工作
經患兒家長(監護人)同意,并簽署知情同意書。抽取患兒及父母外周靜脈血各2ml(EDTA抗凝),參照鹽析法血液DNA提取試劑盒操作要求制備基因組DNA,用Sim-100超微量分光光度計(杭州迅捷生物技術有限公司)測定DNA的OD值。DNA總量>5ug;在分光光度計檢測下OD260/280>1.8且DNA濃度大于50ng/ul,電泳質檢未見DNA降解;一次性無菌、無RNA酶的1.5ml 離心管,容器無破損、泄露,采用封口膜對標本管口進行單獨保護;2-8℃保存,一周內送檢。
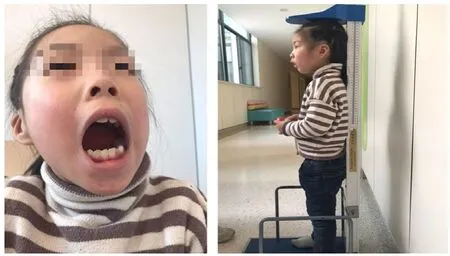
圖1 患兒特殊面容及體格特征
1.2.2 數據分析
測序結果經生物信息學方法進行數據分析。二代測序原始數據(來自杭州嘉檢醫學)的注釋范圍為:每個外顯子的變異,以及外顯子上下游各10 bp內含子中的變異。該基因包檢測區間包括5,177個相關基因,74,566個編碼區總共含有12,424,088個堿基。平均覆蓋深度292+/-185X,大于10X 覆蓋區間占99.8%,大于20X 覆蓋區間占99.6%,檢索HGMD,Pubmed,Clinvar等數據庫,檢索變異相關文獻,分析文獻內容,參考ACMG(美國醫學遺傳學和基因組學學會)變異分類指南,對變異進行分類。
2 結 果
檢測到先證者KMT2A基因雜合變異c.9762_9765delGATT (p. I3255Tfs*14),其父母未攜帶該變異,變異疑似新發,如表1,測序圖見圖2。

表1 基因檢測結果
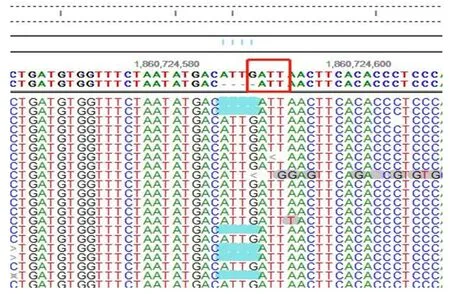
圖2 患兒高精度臨床外顯PLUS檢測KMT2A基因雜合變異測序圖
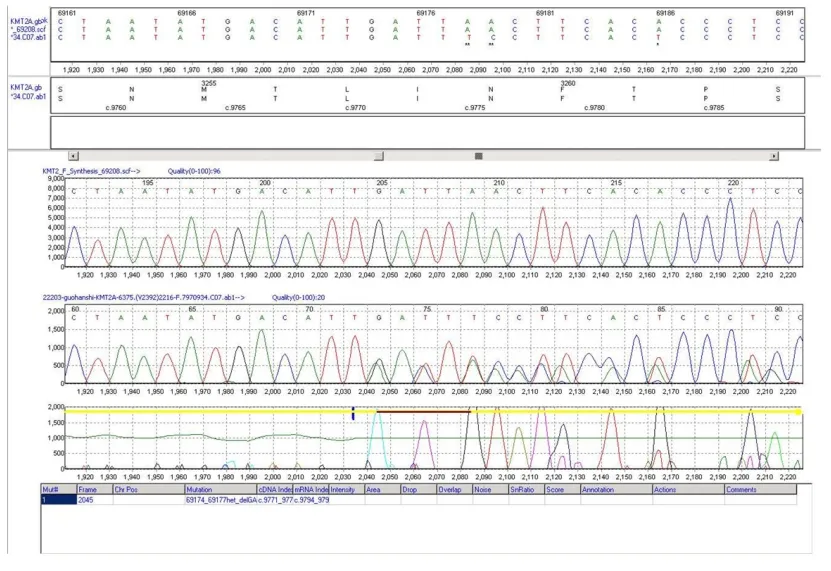
圖3 患兒KMT2A基因雜合變異Sanger 測序圖
變異位點致病性分析:這個變異為新型移碼突變,預測可能會導致蛋白質合成提前出現氨基酸的終止密碼,還沒有在相關臨床病例中被報道過。綜上所述,結合送檢者的臨床表現和家系分析,依據美國ACMG變異分類指南(PMID: 25741868),這個變異為“2類-可能致病”。
3 討 論
Wiedemann-Steiner綜合征,是常染色體顯性遺傳,以新發變異為主。KMT2A基因編碼的蛋白是組蛋白賴氨酸N-甲基轉移酶2A,屬于KMT2家族,其中包括5個編碼(KMT2A-E),已經被證明可以調節染色質期間介導多種發育基因的轉錄胚胎發生和發展[5,6]。目前文獻表述的臨床表現主要為[7,8]身材矮小,發育遲緩,智力異常,語言發育遲緩,特殊面容如長睫毛、濃密的拱形眉毛、寬鼻梁、瞼裂下斜、高腭弓、低位耳等,面部異常如肌張力減退、一側發育不全、上瞼下垂等,其他表現有手指腳趾短小等畸形,肘部多毛,背部多毛,攻擊性行為或自閉癥等。相比以往文獻中臨床表現的記錄,本案例女性患兒符合身材矮小,發育遲緩、智力異常及特殊面容的表現,但未出現肘部或背部多毛等情況,氣質也較溫和,膽小。筆者結合文獻考慮分析:神經發育遲緩作為Wiedemann-Steiner主要特征之一,大多數患者的癥狀為輕度至中度,但在面部外觀方面差異確有顯著,比如毛發,這個案例患兒雖未有此體征,但仍被診斷為兒童無多毛性身材矮小,發育遲緩,由此可看出此基因突變的表現,很可能被臨床醫師低估。
目前全球已報道的KMT2A基因突變引起Wiedemann-Steiner型患者共幾十例[9,10],主要來自歐美地區。由于該病臨床報道較少目前仍無法估計發病率,發病人群無性別差異,無法估計預期壽命。本案例患兒在國內少有報道。Wiedemann-Steiner中描述的臨床特征是廣泛的,對臨床診斷往往具有挑戰性。Wiedemann-Steiner致病性變異陽性患者的臨床表現文獻報道[11],如表2所示的23名患者中分布情況所示。

表2 KMT2A致病性變異型陽性患者中出現在文獻中的臨床特征頻率
經驗總結:在現在和以往基礎研究的結果,提示該病的流行及表型譜可能比預期的要廣泛;因此,臨床醫生對于身材矮小,發育遲緩,面部畸形即使在沒有多毛癥的情況下,是否也應該考慮在兒童中檢測KMT2A基因。基因檢查的意義一方面可以對患者進行的確診,即對特定的異常癥狀或疾病類型臨床診斷的驗證,對疾病的遺傳診斷,提高預后信息;另外可以協助選擇當前臨床最近治療方案;再則對患者家屬患病風險評估。
不足之處:高通量測序分析結果不排除某個基因或基因的某部分存在拷貝數變異的可能性。對于新發變異,不能排除送檢者父母存在體細胞或生殖腺嵌合的可能性。因此建議相關親屬進行相應定點變異檢測,以評估變異來源,疾病相關性及遺傳風險。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
