醋酸卡泊芬凈制備工藝中一個罕見雜質的發現
2019-03-27 13:29:30杜明鏡周陳鋒徐麗華施林峰王麗敏朱益新
安徽化工 2019年6期
杜明鏡,周陳鋒,徐麗華,施林峰,王麗敏,朱益新
(杭州中美華東制藥有限公司,浙江杭州310011)
醋酸卡泊芬凈(默克醫藥公司開發,商品名:Cansidas,英文名:Caspofungin Acetate,CAS:179463-17-3,分子式:C52H88N10O15·2 (CH3COOH) ,分子量:1213.42,結構式見圖1)是2001 年2 月首個上市的棘白菌素類藥物[1],其特點是具有廣譜抗真菌活性,尤其對耐氟康唑和兩性霉素B 念珠菌的抗菌活性較高,對其他念珠菌,如熱帶念珠菌、光滑念珠菌、克柔念珠菌等也有抗菌效果,而且由于其獨特的作用于細胞壁的抗菌機理,不良反應率低[2]。
目前醋酸卡泊芬凈大多采用發酵法得到母核紐莫康定B0,然后通過多步合成得到卡泊芬凈粗品,合成步驟伴隨著多步高壓制備純化,最后得到卡泊芬凈粗品(圖1)[3-4]。高壓制備是當前制藥企業常用的分離純化方法,其特點是分離效果好,周期短,回收率高,但存在填料較貴,對工藝要求高等缺陷,其中填料與工藝的適用性與雜質分析是當前高壓制備的難點之一[5-6]。
在多次的制備實驗過程中,我們偶然發現在卡泊芬凈粗品(即D2與乙二胺的反應產物)純化過程中,柱再生階段時,回收液經儲存后會產生少量無色針狀晶體,過濾得到該晶體,發現其HPLC 出峰與工藝所用物料和中間體都無法匹配。由此,我們對此偶然得到的雜質展開了研究,希望能借該雜質研究進一步豐富對工藝的理解,以降低產品生產的風險。
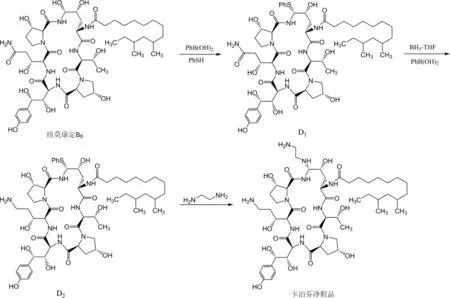
圖1 三步法合成卡泊芬凈粗品
1 實驗試劑和設備清單
主要試劑為食用級乙醇和純化水,其余為自備的檢測試劑。主要設備清單見表1。我們無法得知雜質在其中的出峰時間和歸一化含量。以此作為原料進行養晶實驗。
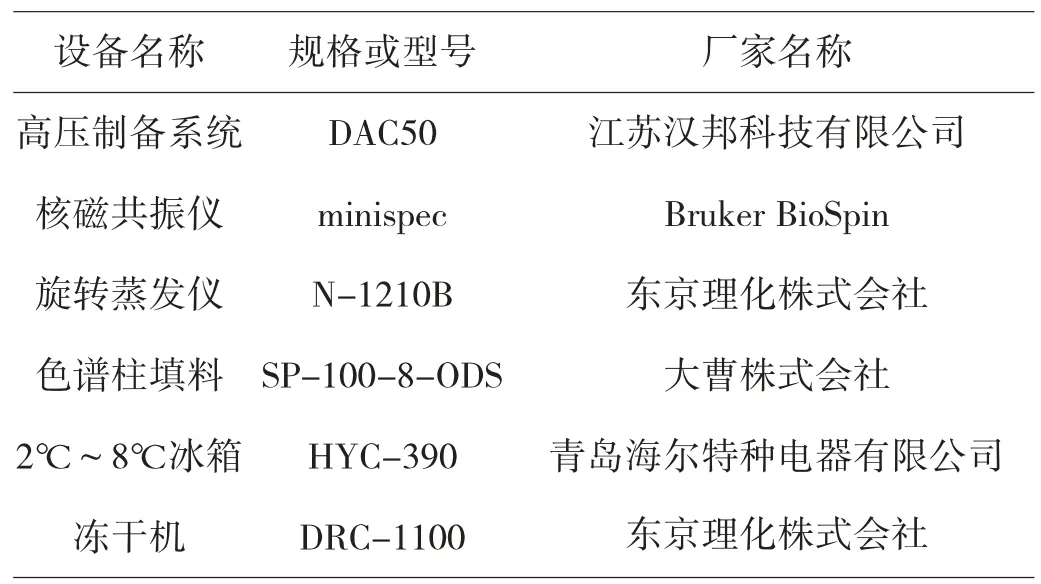
表1 實驗設備清單
2.2 養晶
對上一步得到的再生濃縮液,過濾除去大顆粒,然后將其置于2℃~8℃環境中,靜置12 h,觀察到有規則的無色透明針狀晶體生成,見圖2。
2 實驗步驟與討論
2.1 雜質富集
根據偶然發現,我們認為雜質出現在制備柱再生環節。因此首先將多個中試批次制備再生液合并(再生體系:95% 乙醇),然后用旋蒸儀50℃下旋蒸,得到了高濃度的再生合并液,pH=7.8,呈淡黃色液體狀。由于再生合并液里物質成分復雜,HPLC 峰交錯,分離度不好,

圖2 雜質晶體
2.3 結構鑒定
上一步得到的雜質晶體,過濾,低溫旋干后得到白色固體粉末。HPLC 顯示歸一化純度為93% ,見圖3。然后,進行H NMR 和C NMR 分析,見圖4 和圖5。

圖3 雜質HPLC 分析

圖4 雜質H NMR 圖

圖5 雜質C NMR 圖
由此,結合MS 結果(MW=246.31)和H NMR、C NMR結果,我們解譜分析得出結論:該物質為對甲苯二硫酚(見圖6),其為產品反應液中掉落的苯硫酚殘基在溶液中形成的二聚體,或者也可能是在高壓制備柱的碎填料間形成。
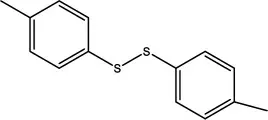
圖6 對甲苯二硫醚結構式
對甲苯二硫醚(英文名稱:1,2-di-p-tolyldisulfane,分子式:C14H14S2,分子量:246.39,CAS: 103-19-5),具有使皮膚過敏和眼睛發炎的危害,并可導致呼吸道發炎,在制藥產業中要注意避免直接吸入,不能直接排入下水道,需隨生產廢液一起進行合法處理和轉移。
2.4 生產工藝改進
由以上結果可知,該雜質產生于D3 制備過程中,可能是在制備母液中形成的,也可能是在高壓的碎填料間形成的,而且該雜質與C18 基質的填料結合牢固,只有在95% 乙醇洗滌時,才會緩慢洗脫出來。因此,我們有意延長了生產工藝中的洗脫步驟,改正常流速洗脫3BV 為高低流速交錯洗脫8BV,最后得到的再生液中HPLC 幾乎沒有出峰,由此降低了產品的雜質風險。
3 結論
在不斷的制備過程中,我們偶然發現了對甲苯二硫醚雜質,并著手進行分離、提取、結構鑒定。實驗結果顯示,在醋酸卡泊芬凈的制備過程中,反應液中或制備填料中會使游離的對甲苯硫酚形成二聚體,牢牢吸附在填料上,只有在高有機相再生時才會緩慢洗脫出來。由此我們改進了制備再生工藝,降低了該雜質帶入產品的質量風險。
對于制藥產品來說,雜質控制是永恒的主題,關注工藝細節,關心生產過程現象,發現雜質并用科學的方法去解決雜質問題,是保障產品質量不斷提升的必備手段。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
山東冶金(2019年6期)2020-01-06 07:45:54
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
世界農藥(2019年2期)2019-07-13 05:55:12
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
石油化工應用(2014年8期)2014-03-11 17:40:03
