銻烯吸附金屬Li原子的密度泛函研究*
2019-03-11 08:55:30欒曉瑋孫建平王凡嵩韋慧蘭胡藝凡
物理學報 2019年2期
關鍵詞:結構
欒曉瑋 孫建平 王凡嵩 韋慧蘭 胡藝凡
(華北電力大學電氣與電子工程學院, 北京 102206)
(2018 年9 月3日收到; 2018 年11 月23日收到修改稿)
銻烯(antimonene)是繼石墨烯和磷烯之后出現的新型二維材料, 在鋰離子電池等領域受到關注. 本文基于第一性原理的密度泛函理論, 計算研究了銻烯對Li原子的吸附特性, 包括Li原子的最穩定吸附構型、吸附密度以及吸附Li原子的擴散路徑. 結果表明: Li原子最穩定的吸附位置位于谷位, 即底層Sb原子之上、頂層三個Sb原子中心位置, 吸附能為1.69 eV, 吸附距離為2.81 ?; 能帶計算發現, 銻烯為帶隙寬度1.08 eV的間接帶隙半導體, 吸附Li原子后費米能級上升進入導帶, 呈現出金屬性; 原子分波態密度分析發現, Sb原子的p電子態和Li原子的p和s電子態形成明顯的共振交疊, 表現出雜化成鍵的特征; 隨著吸附Li原子數量增加, 銻烯晶格結構和電子結構發生較大變化. 通過微動彈性帶方法計算發現, Li原子在銻烯表面的擴散勢壘為0.07 eV,較小的勢壘高度有利于快速充放電過程.
1 引 言
鋰離子電池具有高能量密度、高存儲能量、循環壽命長和低損耗的優點, 在日常電子產品、儲能系統中得到廣泛應用. 隨著鋰離子電池在電動汽車、儲能電池、航空航天以及軍事等相關領域需求的快速增長, 人們對鋰離子電池的可逆性容量、倍率充放電能力和循環穩定性等性能提出了更高的要求[1,2]. 由于鋰離子電池性能很大程度上受到電極材料儲鋰容量和循環速率的制約, 傳統負極材料的儲鋰性能已經接近理論極限. 以石墨烯和磷烯為代表的新型二維材料具有獨特結構和優異的電學性能, 在鋰離子電池負極材料領域具有很好的應用前景, 引起了人們的極大關注. 2004年, 英國科學家Geim[3]采用機械剝離法, 首次獲得能夠在外界條件下穩定存在的石墨烯. 石墨烯是碳的一種同素異形體, 是碳原子以sp2雜化形成的六角蜂窩狀單層薄膜, 層厚約為0.335 nm. 石墨烯具有優異的導電性、較高的機械強度等優點. 由于石墨烯獨特的單層碳原子結構, 使Li離子不僅可以存儲在石墨烯片層的兩側, 還可以存儲在石墨烯片層的邊緣和中間, 其最佳吸附位置是碳六邊形的中心[4]. 其理論比容量為740—780 mA·h/g, 鋰離子容易在其中嵌入和脫嵌, 使得電極電導率較高, 表明石墨烯作為鋰離子電池負極材料具有其優越性[5,6]. 有研究表明, 在石墨烯上摻雜其他元素如硼、氮等元素可以改變石墨烯的電學性能, 且氮摻雜石墨烯作為鋰離子電池負極材料具有穩定的電化學性能和更大的儲鋰能力[7-9]. 磷烯是最新出現的二維半導體材料, 且單層和少層黑磷具有直接帶隙和高的電子遷移率, 其直接帶隙的特性彌補了石墨烯用作半導體器件的不足, 在微電子器件方面有很好的應用前景[10].在鋰離子電池領域, 實驗研究表明, 磷烯包覆的鋰離子負極材料可以提升電池的庫侖效率、容量以及循環穩定性[11,12]. 磷烯放電電位范圍為0.4—1.2 V,鋰離子可以在其中快速定向擴散, 在0.2 C電流中放電, 其比容量達到2786 mA·h/g. 理論計算表明,磷烯帶隙為0.91 eV, 導電性好, 且具有較高的吸附能和較低的擴散勢壘, 其中沿鋸齒形方向的單層磷烯擴散勢壘僅為0.09 eV, 表明其具備快速充放電的潛力[14], 高于基于石墨烯的鋰離子電池負極材料[13-15]. 此外, 吸附過渡金屬元素可以提高磷烯作為鋰離子電池負極材料的儲鋰性能[16]. 但是磷烯直接應用于各種器件較為困難, 主要原因是磷烯在空氣中極易氧化, 結構可在數小時內被破壞. 通過修飾、摻雜、涂覆與復合等方式鈍化和功能化磷烯,可以抑制磷烯的氧化, 在一定程度上提高穩定性并改善性能[17].
繼石墨烯和磷烯之后, 最近出現的二維材料銻烯(antimonene), 由于具有良好的化學穩定性和獨特的物理特性, 在半導體電子器件、鋰離子電池等領域引起廣泛關注[18,19]. 它是一種完全由Sb原子組成的具有皺褶起伏的類石墨烯結構, 是一種新型二維間接帶隙半導體材料, 具有優良的光電子學和自旋電子學性質[20,21]. 目前, 有報道實驗上已經可以通過機械剝離、液相剝離、氣相生長等方法制備化學穩定的銻烯[22]. 在鋰離子電池領域, 人們之前對Sb塊體的研究發現, 其理論嵌鋰容量可達到660 mA·h/g, 嵌鋰電位為0.8 V, 比金屬鋰稍高的電位能夠有效避免枝晶的出現, 提高電池安全性.實驗發現, 塊體銻材料在嵌脫鋰時存在較大的體積膨脹和收縮, 導致電池循環特性變差, 縮短了電池工作壽命. 對于多層和單層銻烯, 隨著厚度減小,由于量子尺寸效應, 其電子結構由金屬型向半導體轉變, 會對鋰原子吸附特性產生重要影響; 此外,隨著尺寸和維度減小, 體積效應會逐漸減弱, 采用銻烯電極材料的電池循環壽命可望得到延長, 比表面積增大, 也有利于提高儲鋰容量. 因此, 開展銻烯的理論和實驗研究具有重要意義. 理論計算方面, 有報道銻烯對于H2O, O2等小分子有較弱的物理吸附, 而對于H, C, N, O, Al等原子吸附較強[23]. 此外, 結構缺陷、襯底和吸附都會對銻烯穩定性和電子結構帶來影響[24-26]. 但是, 目前關于銻烯吸附Li原子的系統研究工作還少見報道. 本文用密度泛函理論計算方法系統研究了銻烯對Li原子的吸附特性, 其中包括Li原子的最穩定吸附構型、吸附密度以及Li原子的擴散路徑, 揭示出銻烯具有作為鋰離子電池負極材料的良好潛力, 這一結果可以為實驗研究提供借鑒. 需要指出的是, 實際應用中, 由于制備難度和團聚效應, 直接采用單層銻烯作為鋰離子電池電極材料存在困難, 更大可能是采用多層銻烯材料. 多層結構當層間距較大時, 嵌入其間的鋰原子作用類似單層吸附; 但當層間距變小時, 加之堆疊方式可能不同, 結構會比較復雜, 需要大量詳細的分析計算, 對多層結構有待進一步研究.
2 計算的細節
文中的計算是采用基于密度泛函理論的VASP軟件包來進行的. 其中, 能量交換關聯能函數采用Perdew-Burke-Ernzerhof (PBE)形式的廣義梯度近似 (general gradient approximate)法, 價電子與離子之間的相互作用勢采用綴加平面波贗勢法(pseudopotential augmented wave)來描述. 結構優化和計算時采用截斷能為500 eV的平面波基組展開, 在布里淵區積分計算時采用了VASP軟件推薦的原點在Γ點的Monkhorst-Pack型網格,k點取值為11 × 11 × 1. 弛豫計算時核離子步收斂精度取為0.01 eV/?, 能量收斂精度取為1 ×10-4eV. 選用4 × 4 × 1銻烯原胞共32個原子,Z方向上真空層取20 ?厚度以避免層間干擾, 并考慮自旋極化作用. 結構優化時, 對所有原子進行弛豫. 原子的吸附能定義為:Ead=Eantimonene+Eatom-Eadsorb, 式中Eatom為單個Li原子的能量,Eantimonene為本征銻烯的能量,Eadsorb為吸附之后整個體系的能量. 根據這個定義,Ead值越大表示吸附能越大, 吸附越穩定.
3 結果與分析
3.1 本征銻烯
為了和吸附Li原子之后的銻烯做對比, 首先對本征銻烯進行計算. 選取4 × 4本征銻烯超晶胞, 如圖1(a)和圖1(b)所示, 褐色圓點表示Sb原子, 可以看出Sb原子分為上下兩層, 呈六邊形周期性分布, 為使立體效果更好, 將俯視圖稍做傾斜.本征銻烯中近鄰Sb原子間距為2.87 ?.
圖1(c)是4 × 4本征銻烯取費米能級EF= 0 eV時的能帶圖, 從圖1(c)可以看出, 銻烯能帶中價帶頂起伏大、色散較明顯, 導帶底變化較平緩. 能帶圖中, 價帶頂落在布里淵區中心G點, 導帶底在G和K點之間, 計算得到的帶隙寬度為1.08 eV,為間接帶隙半導體. 用PBE方法計算存在帶隙偏小的問題, 實際銻烯的帶隙會更大. 圖1(d)是4 ×4本征銻烯態密度圖, 可以看出存在明顯的帶隙,而且態密度分布相對于費米能級不對稱. 以上結果與文獻報道一致[23], 符合一般認識, 表明我們選擇的計算方法和參數設置是可靠的.
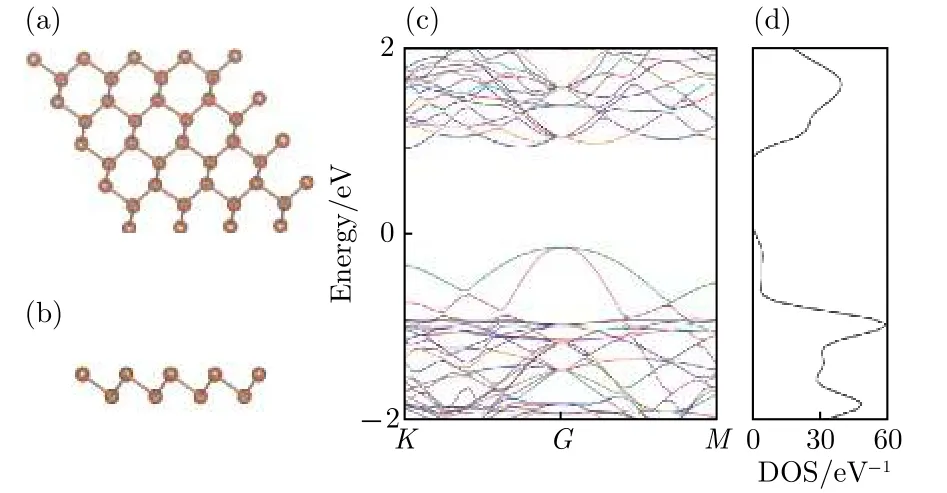
圖1 本征銻烯 (a) 俯視圖; (b) 側視圖; (c) 能帶圖;(d) 態密度圖Fig.1. Pristine antimonene: (a) The top view; (b) the side view; (c) the band structure; (d) the density of states.
3.2 銻烯吸附單個Li原子
為了研究單個Li原子在銻烯上最穩定的吸附構型, 選取4 × 4本征銻烯超晶胞, 吸附一個Li原子, 考慮三種可能的吸附位置: 頂位, 在頂層Sb原子正上方; 谷位, 在頂層三個Sb原子中心, 底層一個Sb原子上方; 橋位, 在頂層兩個Sb原子聯線中心. 先結構優化, 對體系的各個原子進行弛豫. 結果發現橋位上的鋰原子在優化之后回到了谷位的位置, 所以下面我們就谷位和頂位兩種結構進行分析. 兩種初始構型和優化構型分別如圖2、圖3所示, 結構圖中褐色圓點表示Sb原子, 綠色圓點表示Li原子, 為使立體效果更好, 將俯視圖稍微傾斜.
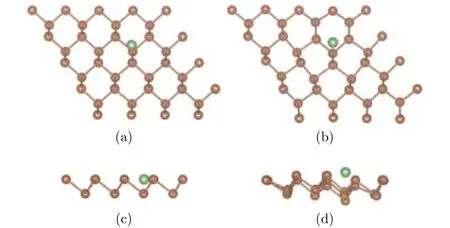
圖2 (a)和(c)分別為谷位吸附結構初始構型的俯視圖和側視圖; (b)和(d)分別為其優化構型的俯視圖和側視圖Fig.2. The antimonene structure of Li adsorbed on vacancy site: (a) The top view before optimization; (b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
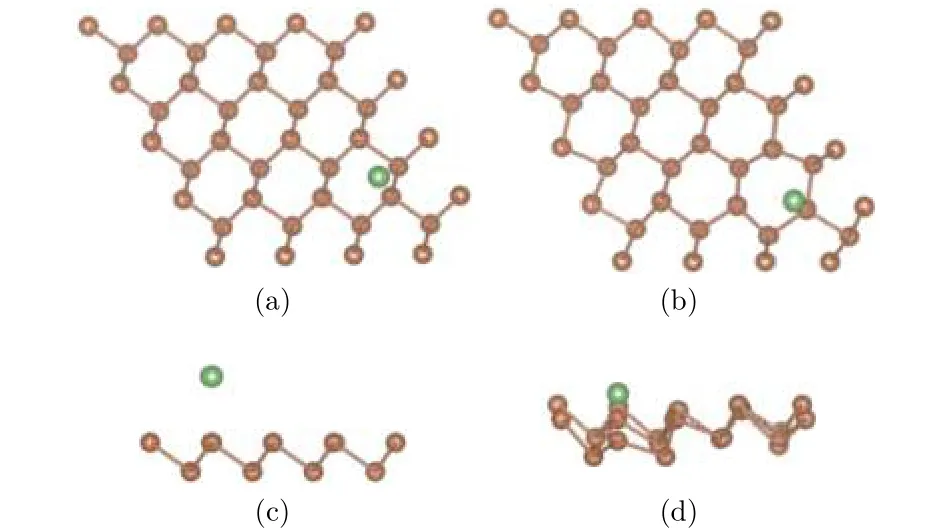
圖3 (a) 和 (c) 分別為頂位吸附結構初始構型的俯視圖和側視圖; (b) 和 (d) 分別為其優化構型的俯視圖和側視圖Fig.3. The antimonene structure of Li adsorbed on top site: (a) The top view before optimization; (b) the top view after optimization; (c) the side view before optimization;(d) the side view after optimization.
通過對比兩種結構的初始構型和優化結果, 可以發現Li原子趨向于吸附在谷位, 也即三個Sb原子形成的中心位置. 從兩種結構的俯視圖來看, 頂位和谷位初始構型的Li原子經過優化后, 位置沒有明顯改變. 而從兩種結構的側視圖來看, 頂位的Li原子相較于初始結構的高度降低, 谷位的Li原子高度相對變化較小.
表1給出了兩種構型的吸附構型、Li原子的吸附能、吸附距離和電荷轉移量. 其中吸附能Ead定義為是Li原子吸附銻烯之后體系的能量,Eantimonene是本征銻烯能量,Eatom是Li原子能量. 可以看出,Li原子吸附的吸附能適中, 屬于化學吸附, 其在頂位的吸附能是1.67 eV, 谷位的吸附能是1.69 eV.吸附能越大, 通常表示吸附后的結構越穩定, 由谷位的吸附能最大可知, 谷位吸附結構最穩定. 吸附穩定性首先可以從吸附原子成鍵的角度進行分析,從成鍵時配位原子的數量來講, 谷位的Li原子可以與周圍三個Sb原子以及下層Sb原子發生相互作用成鍵. 而頂位的Li原子只和一個Sb原子進行相互作用成鍵. 其次, 從優化結構中Li原子和最近鄰的Sb原子的距離可以看出, 谷位吸附距離最短,為 2.81 ?, 頂位吸附距離稍長, 為 2.83 ?, 成鍵距離短通常表示相互作用強, 說明谷位Li原子吸附最穩定. 從Barder電荷轉移也可以進行分析. 由表1可以看出, 吸附過程中, Li原子幾乎完全失去1個價電子, 而鄰近的Sb原子得到電子, 特別是吸附Li原子最近鄰的Sb原子得到較多的電荷. 谷位吸附結構中, 與Li原子最近鄰的Sb原子得到的電荷最多, 為0.32e, 其次是頂位結構, 0.31e, 間接表明了谷位結構中Li原子和Sb原子的相互作用較強.

表1 銻烯吸附Li原子的兩種結構的特性Table 1. Properties of two Li adsorbed antimonene configurations.
由于谷位吸附構型對應于最穩定構型, 所以針對這種構型進行進一步的分析計算. 計算得到的谷位吸附Li原子的銻烯的能帶、總態密度(DOS)和原子分波態密度(PDOS)如圖4所示.
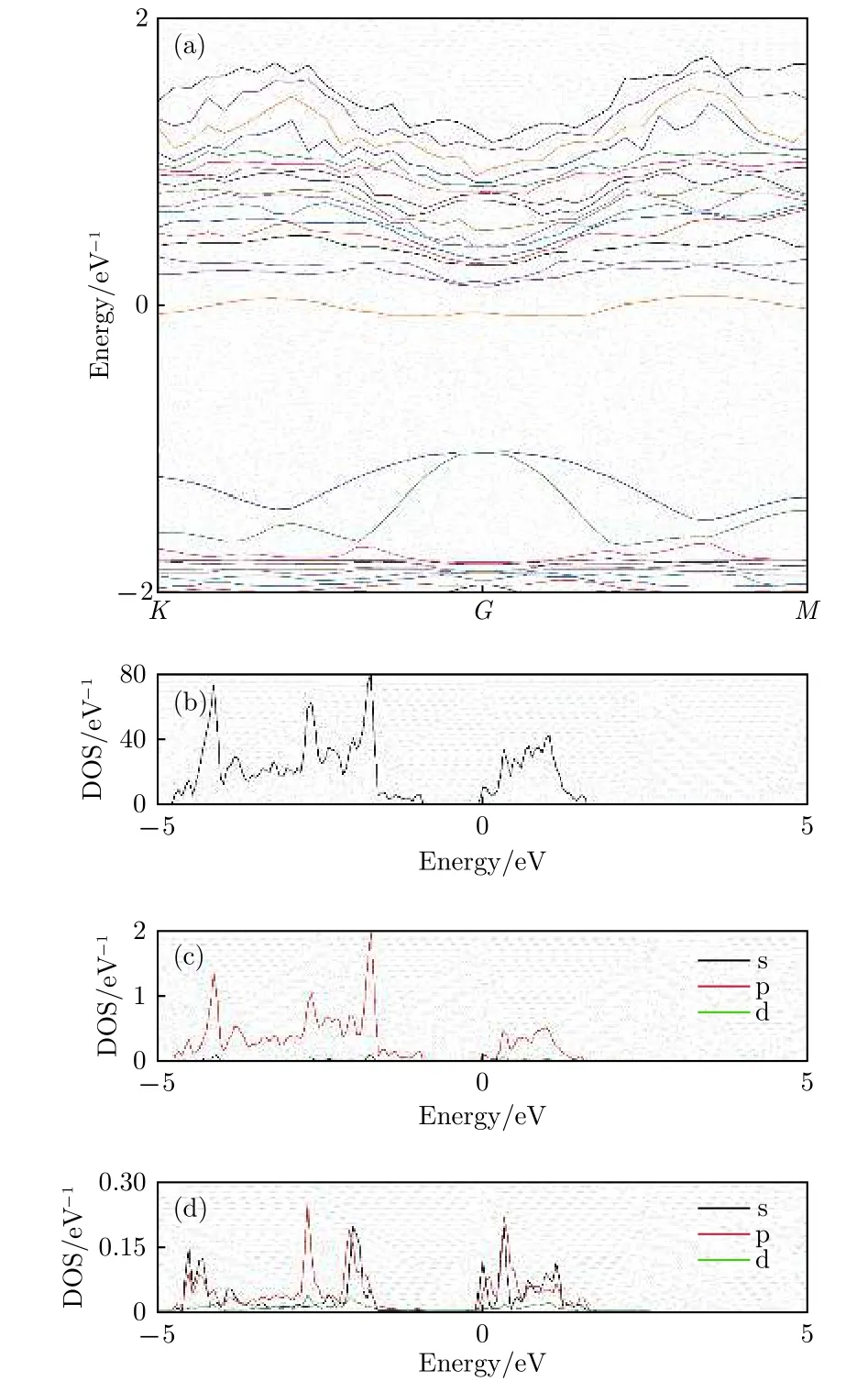
圖4 谷位吸附構型 (a)能帶圖; (b)總態密度;(c) Sb分波態密度; (d) Li分波態密度Fig.4. The band structure and density of Li adsorbed antimonene: (a) The band structure; (b) the total density of states; (c) the partial density of states of Sb atom;(d) the partial density of states view of Li atom.
從圖4(a)與本征銻烯相比較能帶有明顯的改變. 計算得到的谷位吸附結構費米能級在-1.64 eV處, 比本征費米能級有所升高, 以費米能級為零點做圖, 可以看出在EF= 0 eV附近, 帶隙間靠近導帶底附近出現了雜質能帶, 而費米能級穿過雜質能帶, 呈現出N摻雜特征. 相比本征銻烯, 吸附后能帶在價帶頂附近變化不大, 而在導帶底附近分布更密, 起伏更大. 從谷位吸附Li原子的銻烯總態密度也可以清楚看出, 相比于本征銻烯, 費米能級EF=0 eV上移進入導帶. 更進一步, 從Sb原子和Li原子的分波態密度可以看出, 總體上, 主要是Sb原子的p電子態和Li原子的s, p電子態在-5—2 eV區間發生雜化交疊, 產生出一系列共振峰, 表現出Li和Sb原子雜化成鍵的特征. 具體而言, 費米能級處的雜質能帶是由Sb原子的p電子和Li原子的s, p電子交疊形成, 而價帶頂處由于沒有共振峰, 所以能帶不受影響, 這和前面能帶分析的結果一致. 由于費米能級處能帶變化較大, 對體系導電特性的影響顯著, 而導電性的增強對于鋰離子電池電極材料而言顯然是有利的, 有利于減少充放電時的損耗.
圖5給出了頂位吸附Li原子時, 銻烯的總態密度和分波態密度. 從圖5可以看出, 兩種吸附構型對應的總態密度圖相似, 雜化峰位置基本沒變,但相對高度發生了變化; 谷位吸附構型相對于頂位吸附, 在-1—5 eV能量區間, 雜化峰高度有一定程度的提高, 顯示Sb原子和Li原子相互作用增強. 從Sb的PDOS圖中可以看出, 這一變化主要來源于Sb的p電子軌道貢獻; 另外, 對比兩種構型Li原子PDOS中p軌道和s軌道對雜化峰的貢獻, 發現谷位吸附時p軌道在雜化軌道所占比重上升, s軌道相對下降, 這反映出谷位吸附時Li原子和多個Sb原子成鍵的狀態. 態密度分析結果和前面對吸附能、吸附距離和bader電荷分析得到的結果一致.

圖5 頂位吸附結構態密度圖 (a)總態密度; (b) Sb分波態密度; (c) Li分波態密度Fig.5. The density of states of the Li adsorbed structure:(a) The total density of states; (b) the partial density of states of Sb atom; (c) the partial density of states view of Li atom.
圖6給出了Li原子吸附后, 反映吸附結構中各個原子電荷轉移狀況的差分電荷密度圖. 結構圖中褐色圓點表示Sb原子, 綠色圓點表示Li原子,黃色和藍色表示電荷密度高低. 由圖6可以看出,總體上谷位吸附結構中Li原子是失去電子的狀態,而Li原子所失去的電荷不但轉移到了臨近的Sb原子之上, 而且更遠處的Sb原子上也發生了電荷的相互轉移. 綜合前面對態密度的分析, 可以發現, Li原子吸附成鍵, 類似于金屬鍵, 具有非局域的特征.
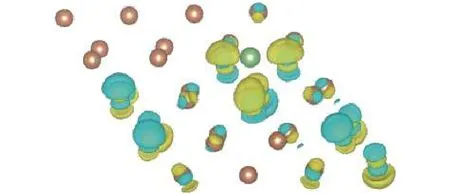
圖6 谷位差分電荷密度Fig.6. The differential charge density of the Li adsorbed structure.
3.3 銻烯吸附多個Li原子
為研究吸附Li原子的密度對整個銻烯構型的影響, 分別構建銻烯吸附3個、6個和9個Li原子的吸附模型, 吸附位置都選擇最穩定的谷位. 圖7、圖8分別是吸附3個和9個Li原子的模型圖, 其中褐色圓點表示Sb原子, 綠色圓點表示Li原子,采用自動優化的方法對所有原子進行弛豫.
由Li原子不同吸附密度的弛豫結果可以看出,銻烯吸附Li原子之后, 由于和Sb原子間的相互作用, 導致自身原子位置發生偏移. 圖7中, 從吸附3個Li原子的結果可以看出, Li原子在水平面內,相對于谷位位置略有偏移, 和Li原子近鄰的Sb原子在水平面內位置變化不大, 底層Sb原子在垂直方向下降, 總體偏移不大. 隨著吸附原子數量增多,如圖8銻烯吸附9個Li原子所示, Li原子吸附的周期性結構一定程度上遭到破壞, 無論是平面方向還是垂直方向, 結構變化較大, 可以認為出現了結構混亂, 這和Li與Sb原子間相互作用比較強有關.
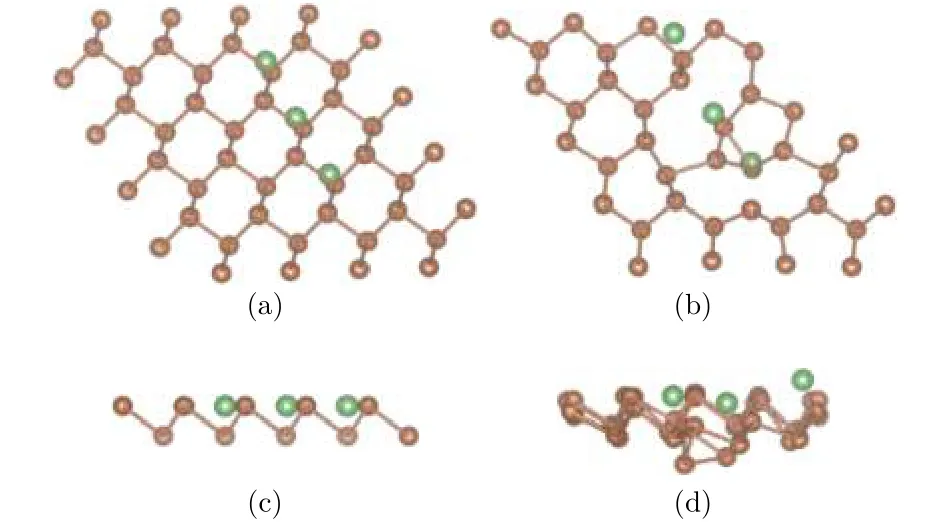
圖7 (a)和(c)分別為銻烯吸附3個Li原子結構初始構型的俯視圖和側視圖; (b)和(d)分別為其優化構型的俯視圖和側視圖Fig.7. The configurations of antimonene adsorbing three lithium atoms: (a) The top view before optimization;(b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
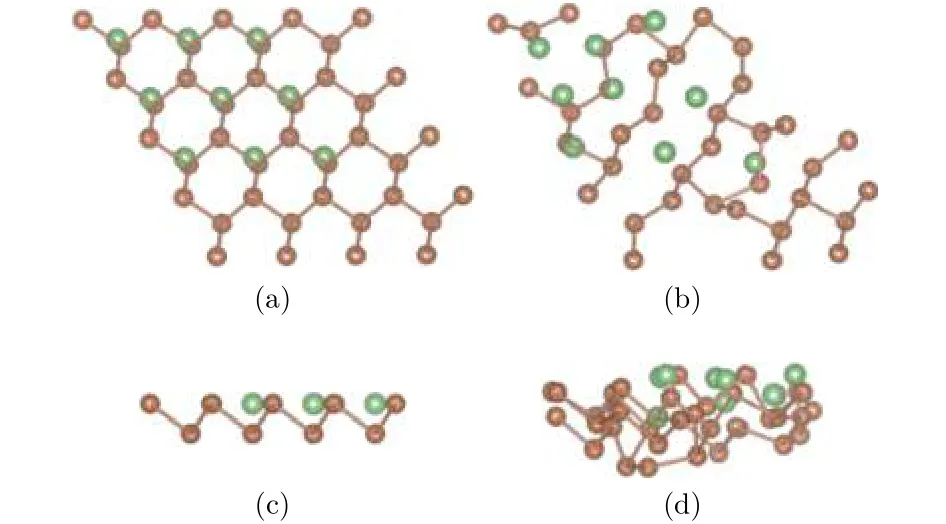
圖8 (a)和(c)分別為銻烯吸附九個Li原子結構初始構型的俯視圖和側視圖; (b)和(d)分別為其優化構型的俯視圖和側視圖Fig.8. The configurations of antimonene adsorbing nine lithium atoms: (a) The top view before optimization;(b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
我們定量計算的不同數量的Li原子吸附構型中, 平均每個Li原子的平均吸附能Ead定義為:其中Eatom為Li原子的能量,n為吸附Li原子的數量. 銻烯吸附Li原子數量為1, 3, 6, 9時,原子平均吸附能分別為1.69, 1.15, 0.94, 0.97 eV. 可以看出,隨著銻烯吸附Li原子數量從1個增大到6個, 原子平均吸附能從1.69 eV逐漸下降到0.94 eV, 這符合化學吸附的一般規律. 一般認為, 隨著吸附密度增大, 吸附Li原子間的相互作用增強, 會引起Li原子和臨近Sb原子間相互作用的削弱. 另一方面,可以看到, 這一變化趨勢隨著吸附密度的進一步增大而遭到破壞, 當吸附數量達到9個時, 平均吸附能增大到0.97 eV, 而這應該是Li原子吸附引起銻烯結構顯著改變引起的.
圖9給出了銻烯吸附3個Li原子的態密度,和圖5中吸附單個Li原子態密度比較, 可以看出,它們有少量不同之處, 總體差別不大. 相似之處在于, 如圖9(a)和圖9(b)所示, 吸附體系總態密度和Sb原子分波態密度中峰的位置、形狀和相對高度大致未變, 顯示體系總體電子結構未變; 差別之處是, 如圖9(c)中所示, 單個Li原子吸附時, Li原子分波態密度在-5—2.5 eV能量范圍呈現為多個分立的共振峰; 3個Li原子吸附時, 靠近-5 eV低能端的峰消失, 變成連續能帶, 這一變化主要是由于Li原子間的相互作用產生的. 總態密度圖相差不大, 說明吸附Li原子對銻烯整體電學特性的影響不大.
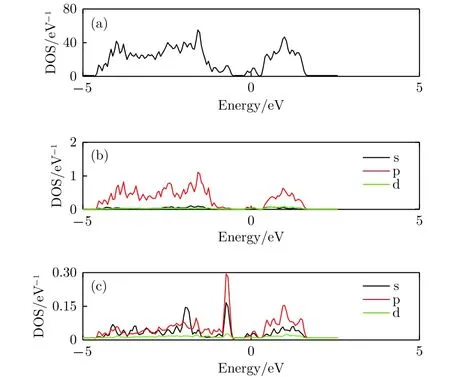
圖9 銻烯吸附三個Li原子態密度圖 (a) 吸附結構的總態密度圖; (b) Sb原子的分波態密度; (c) Li原子的分波態密度Fig.9. The density of states of antimonene adsorbing three lithium atoms: (a) The total density of states of the structure; (b) the partial density of states of Sb atom;(c) the partial density of states view of Li atom.
圖10給出了銻烯吸附九個Li原子的態密度圖, 和圖5相比, 銻烯吸附九個Li原子的態密度圖變化較大. 從圖10(a)和圖10(b)中可以看出, 銻烯的總態密度和Sb原子分波態密度的形狀發生了改變, 整體更加平緩. 此外, 帶隙變窄幾乎消失, 呈現典型的金屬性. 由圖10(c)也可以看出, 原來Li原子分立的共振峰很大程度上消失, 變成連續分布的能帶, 說明隨著吸附原子數量的增加, 銻烯電子結構有逐漸從孤立原子吸附狀態向合金化轉變的趨勢. 綜合以上分析可以看出, 隨著吸附Li原子數量的增加, 一方面對銻烯的晶體結構造成較大擾亂, 另一方面也會引起電子結構的改變. 對于單層銻烯而言, 要達到最大儲鋰密度, 穩定性應該是需要考慮的重要因素. 實際應用中, 結構更加穩定的多層銻烯模型應該更符合實際狀況, 有待下一步研究.
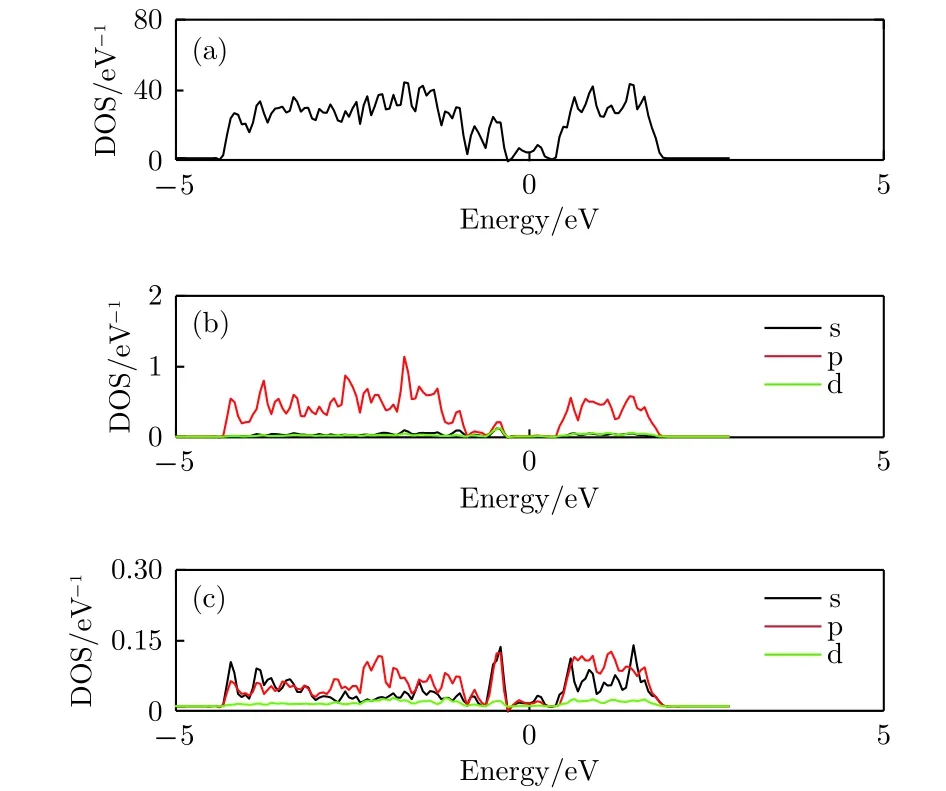
圖10 銻烯吸附九個Li原子態密度圖 (a) 吸附體系的總態密度圖; (b) 體系中Sb原子的分波態密度; (c) 體系中Li原子的分波態密度Fig.10. The density of states of antimonene adsorbing nine lithium atoms: (a) The total density of states of the structure ; (b) the partial density of states of Sb atom;(c) the partial density of states view of Li atom.
3.4 吸附Li原子的遷移
Li原子在鋰離子電池電極材料內的擴散難易直接影響電池的充放電速率, 為了弄清楚吸附Li原子在銻烯表面的擴散遷移過程, 即從穩定吸附位置遷移到鄰近的另一個穩定位置的路徑, 采用VASP自帶的微動彈性帶(NEB)過渡態搜索的方法進行了計算研究. 首先設定初態和終態兩個穩定位置, 然后選取插點數量為三個, 通過計算得到從初態到終態的遷移路徑上結構和各個狀態的能量變化.
圖11結構圖中褐色表示Sb原子, 綠色表示Li原子. 圖11中繪出的5個Li原子表示了Li原子的擴散遷移路徑, 最左和最右的Li原子分別表示初態和終態的穩定吸附位置, 記為狀態1和狀態5, 中間三個對應于擴散中間狀態2, 3, 4, 其中狀態3位于擴散路徑的中點. 圖12給出了遷移過程中從初態到終態體系能量的變化. 可以看出, 狀態3能量明顯最高, 而且恰好位于路徑中點, 根據對稱性判斷, 狀態3應該對應于勢能面鞍點, 稱為過渡態. 通常規定初態和過渡態的能量差為擴散勢壘, 勢壘高度決定了擴散發生的難易程度, 勢壘低,則從一個穩定態遷移到另一個穩定態越容易, 外加電場條件下遷移發生也越快, 反之則越困難越慢.從圖11中數據可以計算得到擴散勢壘高度Eb=0.07 eV, 鋰離子電池充放電的快慢程度常用倍率特性描述, 大倍率充放電能力在很多場合非常重要. 其中電極材料對鋰離子的擴散能力是影響充放電倍率的一個重要因素, 擴散勢壘低說明擴散和遷移更加迅速, 較小的勢壘高度表明, 銻烯用作電極材料有利于發生快速充放電.
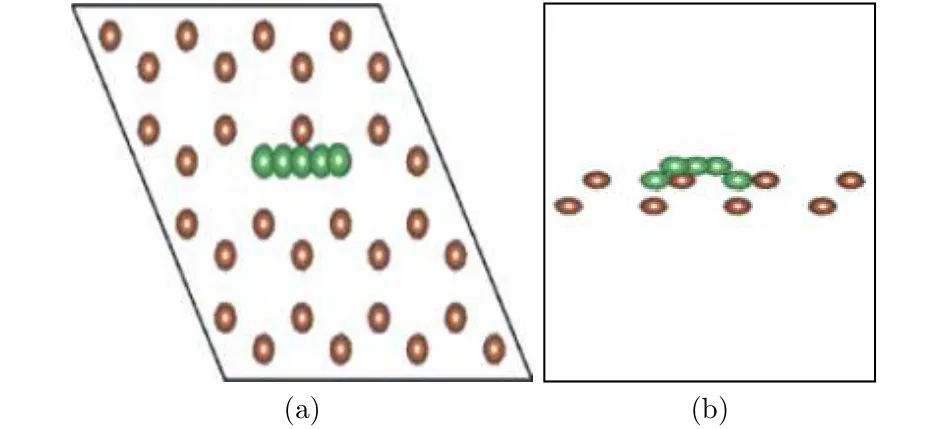
圖11 Li原子擴散遷移路徑 (a) 俯視圖; (b) 側視圖Fig.11. The Li diffusion path on antimonene: (a) The top view; (b) the side view.
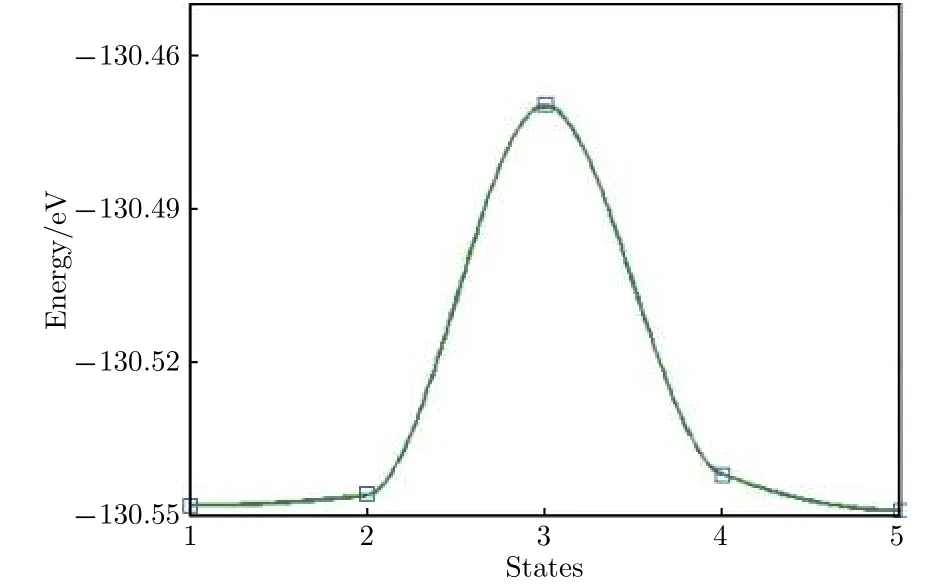
圖12 Li原子擴散遷移勢壘Fig.12. The diffusion energy barrier of Li atom.
4 結 論
銻烯是繼石墨烯和磷烯之后, 實驗中最新制備獲得的二維半導體材料, 由于具有很好的穩定性和獨特的電學特性, 在半導體器件和鋰離子電池等領域引起人們的廣泛研究興趣. 本文采用第一性原理的密度泛函方法, 通過理論模擬計算, 系統地研究了Li原子在單層銻烯上的吸附特性; 發現最穩定的吸附構型是谷位, 吸附能為1.69 eV, 吸附距離為2.81 ?. 電子結構計算發現, 銻烯為帶隙寬度1.08 eV的間接帶隙半導體, 吸附Li原子后費米能級上升穿過雜質能帶, 呈現出金屬性; 隨著吸附Li原子數量的增加, 單層銻烯晶體結構和電子結構都出現了較大改變. 實際應用中, 采用多層銻烯結構或復合結構(如石墨烯/銻烯), 有望提高穩定性從而獲得更高的儲鋰密度; 通過NEB過渡態搜索的計算方法研究Li原子遷移路徑, 發現Li原子擴散勢壘為0.07 eV. Li原子在銻烯上的遷移勢壘較小, 有利于快速充放電過程. 綜上所述, 銻烯有作為鋰離子電池負極材料的優良潛力, 本文研究結果可為相關實驗研究提供理論參考.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
