基于氮化碳復合材料磁性固相萃取結合高效液相色譜法測定尿液中3種羥基多環芳烴
2019-03-08 09:57:46念琪循劉園滿王曼曼
色譜 2019年3期
念琪循, 劉園滿, 孫 冰, 王曼曼
(華北理工大學公共衛生學院, 河北 唐山 063210)
多環芳烴(polycyclic aromatic hydrocarbons, PAHs)是一類由木材、煤炭和煙草等有機物不完全燃燒產生的持久性有機污染物(persistent organic pollutants, POPs)。研究表明,長期暴露在PAHs下可增加人體罹患肺癌、膀胱癌和乳腺癌等癌癥的發病率[1]。PAHs廣泛分布在大氣、土壤、水體和食品等介質中,僅靠外暴露劑量不能準確評價PAHs對健康的影響。羥基多環芳烴(hydroxylated PAHs, OH-PAHs)是PAHs經人體代謝后主要由尿液排出的一類生物標志物,常作為PAHs的內暴露指標用于全面評價PAHs的暴露水平[2]。
分析尿液中OH-PAHs時,由于尿液樣品組分復雜,且OH-PAHs在尿液中含量較低,因此需要首先對尿液樣品進行前處理以達到凈化和富集的目的。目前,尿液中OH-PAHs的前處理通常采用液液萃取法(liquid-liquid extraction, LLE)和固相萃取法(solid-phase extraction, SPE)[2-5]。
磁性固相萃取(magnetic solid-phase extraction, MSPE)是一種以磁性或可磁化的材料作為吸附劑、僅依靠施加外部磁場將目標分析物與樣品基質分離的前處理技術。相較常規的液液萃取法和固相萃取法,吸附劑比表面積大,擴散距離短,只需要使用少量的吸附劑和較短的平衡時間就能實現低濃度目標物的有效萃取,已成功應用于環境、食品和醫藥分析等領域[6-8]。MSPE吸附劑由磁源(通常為Fe3O4)和非磁功能性材料兩部分組成,前者提供與外部磁場相互作用的磁性功能,后者為吸附目標分析物提供作用位點。目前,基于石墨烯、碳納米管、金屬有機骨架等功能性材料作為MSPE吸附劑的工作已有報道[9,10],新型MSPE吸附劑材料的開發仍是研究的熱點。
氮化碳(carbon nitride, g-C3N4)材料是一種僅由C和N兩種元素組成,具有二維蜂窩狀晶格結構的新型納米材料。該材料化學和熱穩定性良好、比表面積大,另外,其結構中富N功能基團及電子離域特性使得它能夠和一些離子或分子產生絡合、疏水、π-π鍵、氫鍵、靜電力等相互作用,因此有望成為一種理想的吸附分離材料[11]。目前,已有文獻[12]報道將g-C3N4材料與Fe3O4通過物理研磨的方式制備得到磁性g-C3N4材料,但通常物理研磨獲得的復合材料穩定性欠佳,實際應用有一定的局限性。本研究利用溶劑熱法構筑了磁性g-C3N4材料,并結合高效液相色譜-熒光檢測,建立了一種簡單、快速、高效的人尿液中3種OH-PAHs的分析方法。
1 實驗部分
1.1 儀器和試劑
Agilent 1200高效液相色譜儀-熒光檢測器(HPLC-FLD,美國Agilent公司); FEI JEM-2800F聚焦離子束/場發射掃描電子顯微鏡(SEM,美國FEI公司); Brucker D8 VENTURE單晶X射線衍射儀(XRD,德國布魯克公司); LDJ 9 600-1振動樣品磁強計(VSM,美國LDJ公司); Micromeritics ASAP 2460全自動快速比表面積及孔隙分析儀(美國麥克公司)。
所用試劑除特別說明外均為分析純。尿素和乙二醇(EG)購自上海阿拉丁生化科技股份有限公司;六水合三氯化鐵(FeCl356H2O)、三水合乙酸鈉(NaOAc53H2O)、乙酸和無水乙醇購自天津市光復精細化工研究所;β-葡萄糖醛酸酶(≥105unites/mL)購自上海市安譜實驗科技有限公司;甲醇(MeOH)、乙腈(ACN)、乙酸乙酯和丙酮為色譜純,均購自賽默飛世爾科技有限公司(美國)。超純水購自杭州娃哈哈集團有限公司。
1-羥基菲(1-OHPhe,純度98.0%)、3-羥基菲(3-OHPhe,純度98.0%)和1-羥基芘(1-OHPyr,純度98.0%)購自德國Dr. Ehrenstorfer公司,使用MeOH配制以上待測物的儲備液,質量濃度為500 mg/L,于4 ℃下避光保存。將儲備液用MeOH稀釋成質量濃度為1 mg/L的標準工作液。
1.2 磁性g-C3N4的制備
參照Dong等[13]的方法制備g-C3N4材料,將10 g尿素置于箱式電阻爐中,以15 ℃/min的速率從25 ℃升溫至550 ℃,維持4 h,待電阻爐冷卻至室溫后,得到g-C3N4材料。
將270 mg g-C3N4材料經超聲均勻分散在一定體積的EG中,加入270 mg FeCl356H2O和700 mg NaOAc53H2O,劇烈攪拌30 min,將所得混合液轉移至事先預熱的反應釜中,于200 ℃下反應12 h,收集反應產物,使用超純水和無水乙醇反復洗滌,真空干燥,得到磁性g-C3N4材料(見圖1a)。
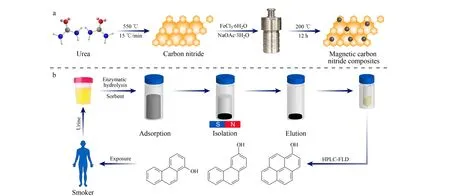
圖 1 (a)磁性氮化碳(g-C3N4)材料的制備和(b)磁性固相萃取流程圖Fig. 1 Flow chart for (a) the preparation of the magnetic carbon nitride (g-C3N4) composites and (b) the magnetic solid-phase extraction
1.3 尿液樣品的制備
采集吸煙志愿者的晨尿,于-20 ℃下冷凍保存。使用時將尿液樣品于室溫下自然解凍并充分混勻,取5.0 mL于離心管中,加入10 μLβ-葡萄糖醛酸酶和5.0 mL 0.5 mol/L乙酸-乙酸鈉緩沖溶液(pH 5),于37 ℃避光水解過夜。將水解后的尿樣于1 500 r/min離心10 min,取上清液于4 ℃儲存待用。
1.4 磁性固相萃取流程
準確稱取4 mg磁性g-C3N4材料,加入2 mL酶解后的尿液樣品中,萃取3 min后,利用磁鐵將吸附劑與尿液樣品分離,棄去上清液。采用0.5 mL丙酮對目標物進行洗脫,充分振蕩3 min后,利用磁鐵提取洗脫液,重復兩次后合并洗脫液,氮吹至干,使用甲醇定容至0.1 mL,待測(見圖1b)。
1.5 色譜分離條件
利用Agilent PAH柱(250 mm×4.6 mm, 5 μm,美國Agilent公司)對3種OH-PAHs進行分離,流動相為ACN和水的混合液。梯度洗脫程序為:0~8 min, 55%ACN; 8~10 min, 55% ACN~65%ACN; 10~16 min, 65%ACN。流速為1 mL/min,進樣體積為20 μL。3種OH-PAHs的激發(Ex)/發射(Em)波長分別為:284/383 nm(1-OHPhe)、250/360 nm(3-OHPhe)、242/396 nm(1-OHPyr)。
2 結果與討論
2.1 磁性g-C3N4的表征
通過SEM對復合材料的表觀形貌進行表征,如圖2a所示,Fe3O4均勻包覆在g-C3N4材料表面;利用XRD對材料的晶體結構進行表征,由圖2b可知,復合材料同時具有Fe3O4的衍射峰和g-C3N4材料的特征衍射峰,由以上結果可知,Fe3O4與g-C3N4材料復合成功。經磁強度(見圖2c)和比表面積分析后可知,磁性g-C3N4材料的磁化飽和度值為36.4 emu/g,比表面積達到113.7 m2/g,滿足磁性固相萃取的需求。
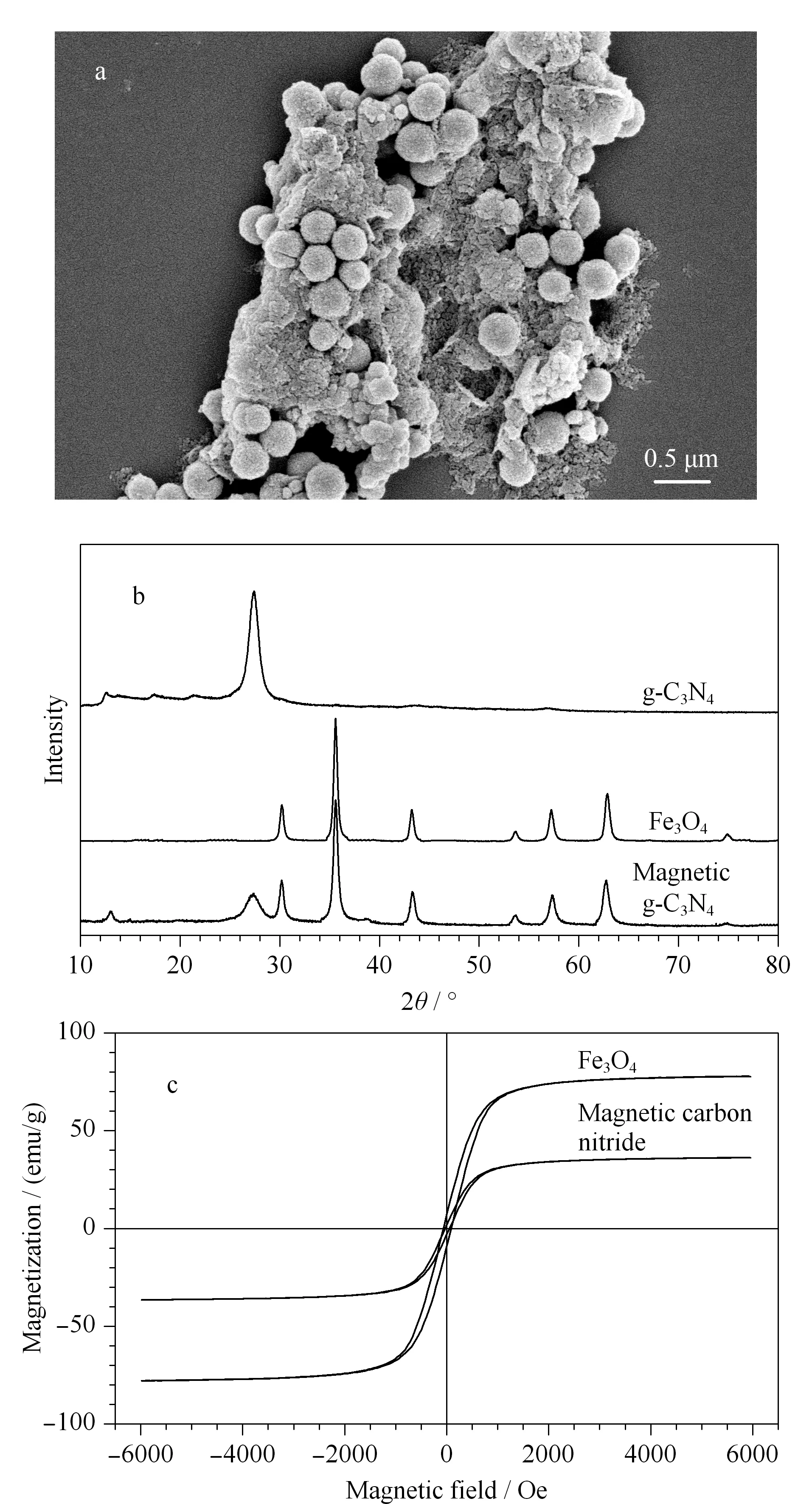
圖 2 磁性g-C3N4材料的(a)掃描電子顯微鏡圖(放大倍數20000)、(b)X射線衍射圖和(c)磁滯曲線圖Fig. 2 (a) SEM image (magnification of 20000), (b) X-ray diffraction (XRD) patterns and (c) magnetization curve obtained for the magnetic g-C3N4 composites
2.2 磁性固相萃取條件的優化
吸附和洗脫條件是決定磁性固相萃取方法準確度的關鍵因素。本研究考察了吸附劑用量、吸附時間、洗脫溶劑的種類和體積(單次洗脫體積×洗脫次數)對目標物萃取效率的影響。使用2 mL 濃度均為40 μg/L的3種OH-PAHs水溶液進行優化試驗,所有試驗平行測定3次。
在固定吸附時間為3 min的條件下,考察吸附劑用量為0.5~5 mg時,對3種目標物吸附效率的影響。如圖3a所示,當吸附劑用量為0.5~2 mg時,1-OHPyr的吸附效率達到最大,1-OHPhe和3-OHPhe的吸附效率逐漸增加;當繼續增加吸附劑用量至4 mg時,1-OHPyr的吸附效率無顯著變化,1-OHPhe和3-OHPhe的吸附效率均達到最大值。繼續增加吸附劑用量至5 mg, 3種OH-PAHs的吸附效率均不再變化,故選用4 mg為最優吸附劑用量。
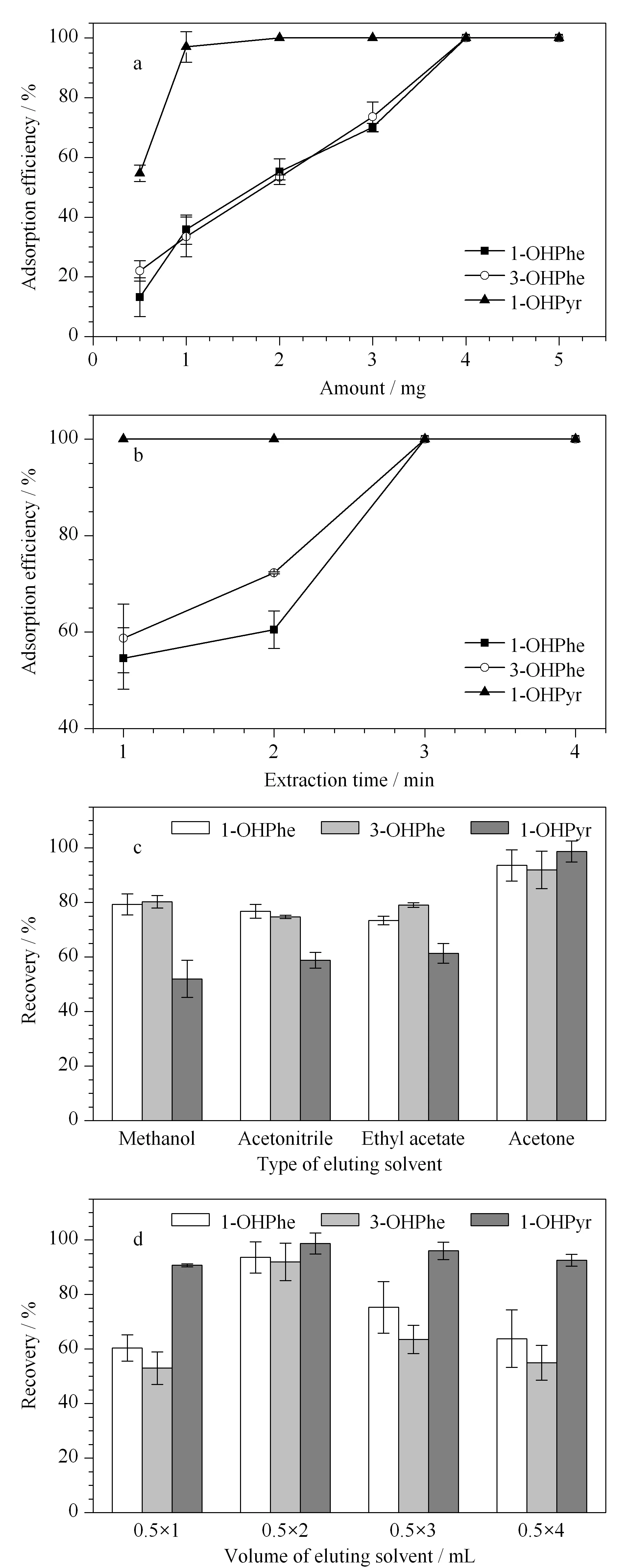
圖 3 (a)吸附劑用量、(b)吸附時間、(c)洗脫溶劑和 (d)洗脫體積(單次洗脫體積×洗脫次數)對3種OH-PAHs萃取效率的影響(n=3) Fig. 3 Effects of the (a) amount of sorbent, (b) extraction time, (c) eluting solvent, and (d) eluting volume (single eluting volume×eluting times) on the extraction efficiencies of the three OH-PAHs (n=3)
固定吸附劑用量為4 mg,考察吸附時間為1~4 min對目標物吸附效率的影響。如圖3b所示,當吸附時間為1 min時,1-OHPyr即達到吸附平衡,1-OHPhe和3-OHPhe的吸附效率仍在增加;繼續增加吸附時間到3 min, 1-OHPyr吸附效率無明顯變化,1-OHPhe和3-OHPhe均達到吸附平衡。繼續增加吸附時間至4 min后,3種OH-PAHs的吸附效率均無顯著變化,故選用3 min作為吸附時間,同時說明本方法可以快速吸附樣品中的3種OH-PAHs。
在最優的上樣條件下,考察MeOH、ACN、丙酮和乙酸乙酯作為洗脫溶劑對目標物回收率的影響。如圖3c所示,洗脫溶劑為丙酮時3種目標物的回收率最佳。同時考察了洗脫溶劑的體積(單次洗脫體積×洗脫次數)對目標物回收率的影響,如圖3d所示,洗脫體積從0.5 mL×1次增加至0.5 mL×2次時,3種目標物的回收率增加,當繼續增加洗脫體積至0.5 mL×3次和0.5 mL×4次時,目標物的回收率下降,這是由于洗脫溶劑體積過大時,氮吹時間延長導致目標物損失,因此選用0.5 mL×2次的丙酮作為最佳洗脫條件。
2.3 方法驗證
2.3.1線性范圍、檢出限和定量限
采用質量濃度為0.25~250 μg/L的OH-PAHs標準溶液進行分析,建立標準曲線。如表1所示,本方法在0.25~250 μg/L范圍內對3種OH-PAHs均表現出良好的線性關系,相關系數(r)均為0.999。以信噪比為S/N=3和S/N=10計算方法的檢出限(LODs)和定量限(LOQs),分別為0.08和0.25 μg/L。結果表明本方法具有良好的靈敏度。
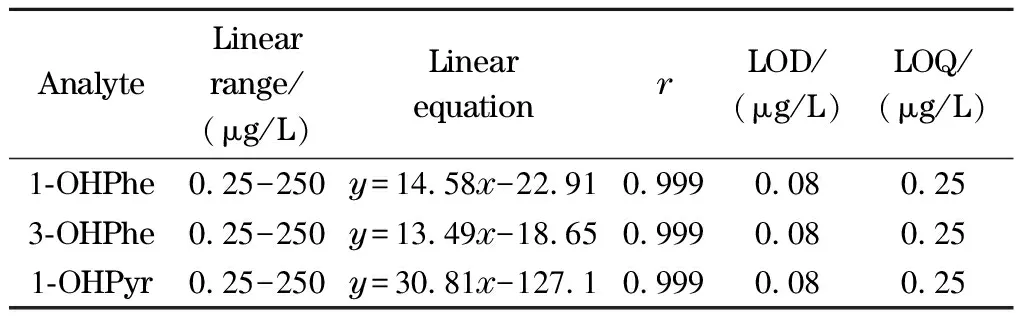
表 1 本方法的線性范圍、線性方程、相關系數、檢出限和定量限Table 1 Linear ranges, linear equations, correlationcoefficients (r), LODs and LOQs of this method
y: peak area;x: mass concentration, μg/L.
2.3.2回收率和精密度
對同一尿液樣品,將1-OHPhe、3-OHPhe和1-OHPyr標準品分別添加至0.5 、1.25 和2.5 μg/L 3個水平 ,經本方法前處理后,計算樣品的加標回收率,以日內和日間分別重復3次測定得到的色譜峰面積的相對標準偏差(RSD)計算精密度,結果如表2所示,方法對3種OH-PAHs的回收率為90.1%~102%,日內精密度為1.5%~7.7%,日間精密度為2.2%~8.7%,表明本方法具有良好的準確度和重復性。
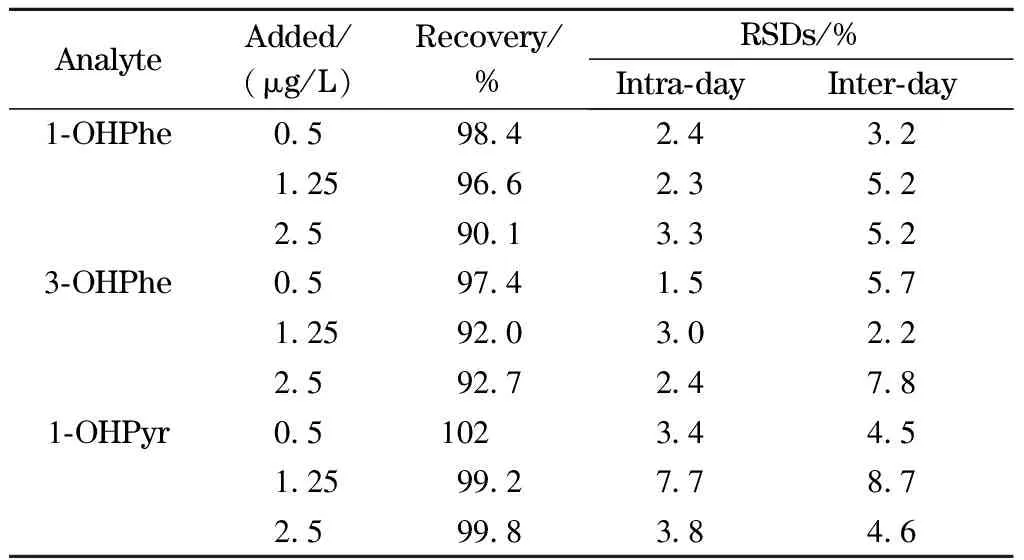
表 2 本方法的回收率和精密度(n=3)Table 2 Recoveries and precisions of this method (n=3)
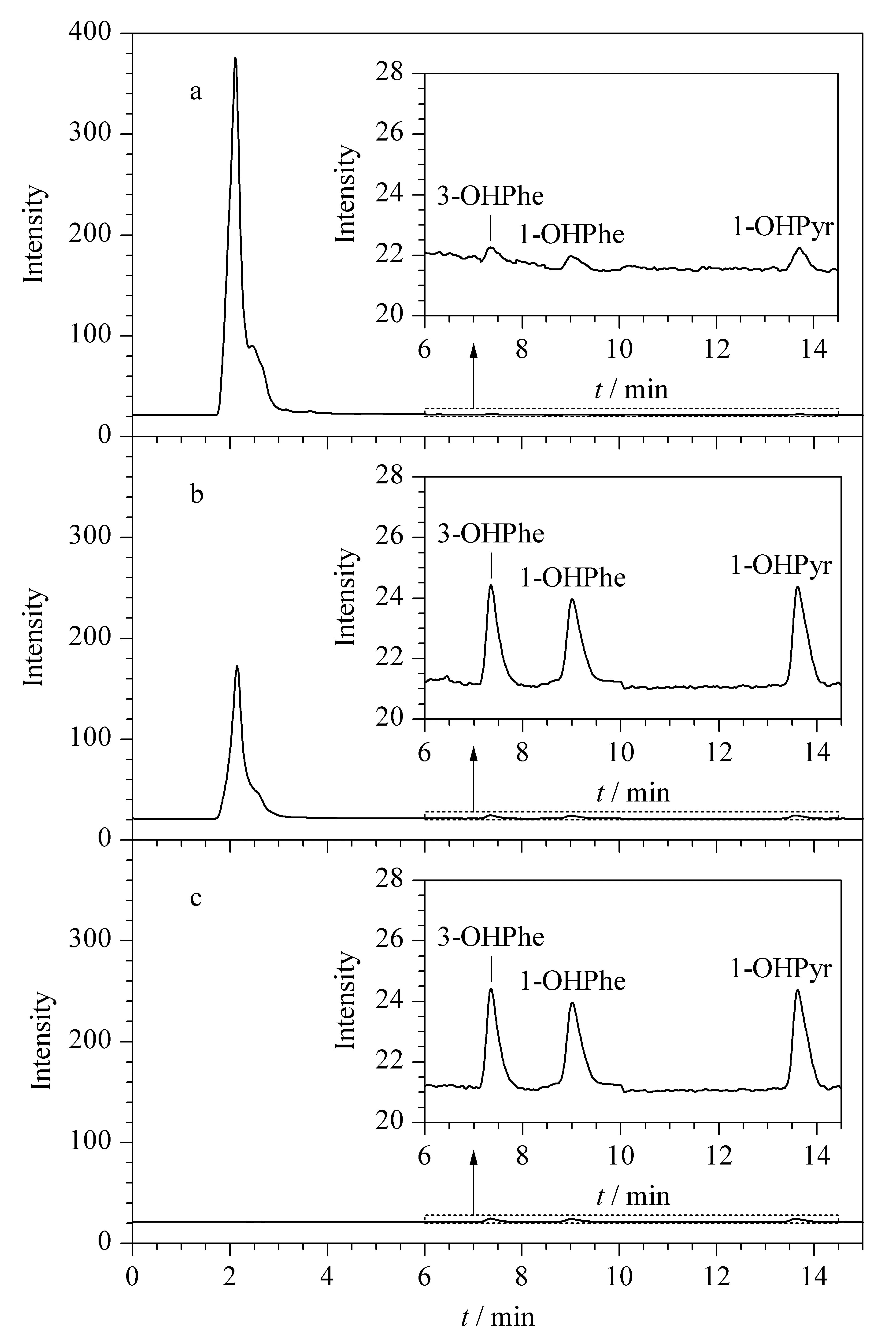
圖 4 加標3種OH-PAHs(均為2.5 μg/L)的尿液樣品 酶解后(a)直接進樣、(b)經本方法前處理后進樣及(c)OH-PAHs標準溶液色譜圖 Fig. 4 Chromatograms of the enzymatic hydrolyzed urine sample (spiked with 2.5 μg/L of each OH-PAH) (a) with direct analysis, (b) pretreated by this method before the analysis, and (c) the standard solution of OH-PAHs
2.3.3凈化和富集效果
在最優條件下,結合HPLC-FLD分析,將加標3種OH-PAHs(質量濃度均為2.5 μg/L)的尿液樣品酶解,經本方法富集凈化前后分別進樣分析。圖4a為直接將尿液樣品進行分析的結果,雜質峰明顯,3種OH-PAHs均有檢出,但樣品峰響應較低;而經過本法固相萃取后,如圖4b所示,雜質峰明顯降低,在對應OH-PAHs出峰位置(見圖4c)均檢出樣品峰且響應高于直接進樣的結果,說明該方法能夠有效去除尿液中的雜質,富集OH-PAHs,當酶解后的尿液上樣體積為2 mL時,對OH-PAHs的富集倍數為10。
2.4 實際樣品分析
利用本方法對4名吸煙志愿者的尿液樣品進行分析。如表3所示,有2例樣品中檢出1-OHPhe,質量濃度分別為(1.65±0.04)和(1.75±0.02) μg/L;有3例樣品檢出3-OHPhe,質量濃度為(1.89±0.05)~(2.26±0.10) μg/L;全部樣品均檢出1-OHPyr,質量濃度為(4.24±0.02)~(4.46±0.01) μg/L。為了進一步說明本方法的可行性,對4例實際尿液進行加標回收試驗,加標濃度為2.5 μg/L,對尿液中的OH-PAHs的回收率為90.7%~108%, RSD≤8.2%,方法準確度和精密度令人滿意。
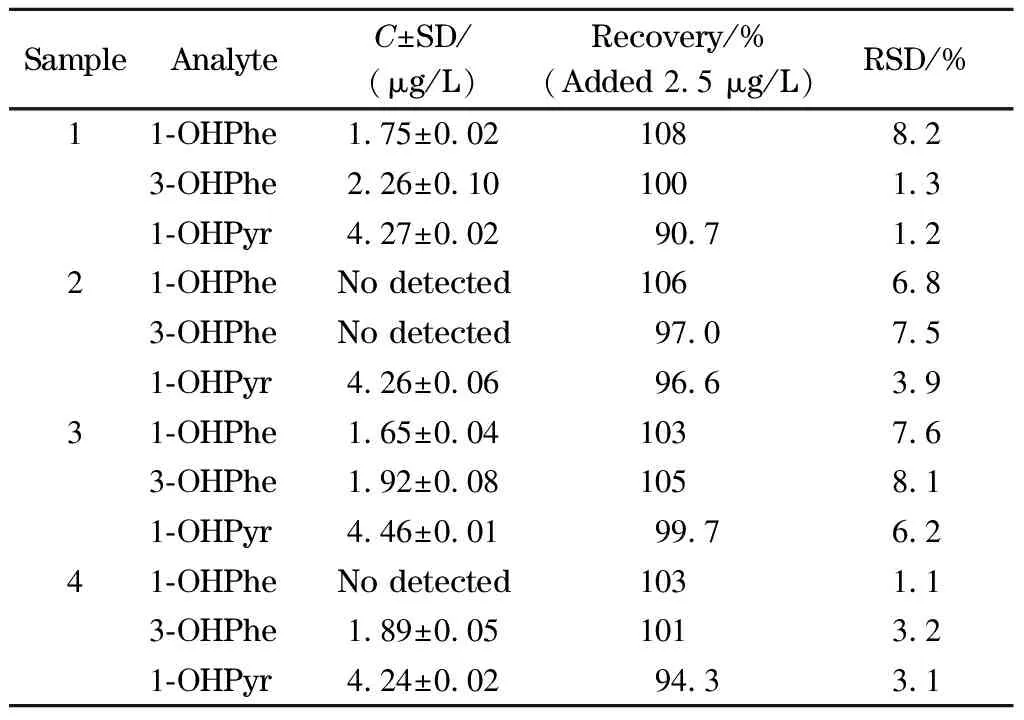
表 3 吸煙志愿者尿液中的OH-PAHs分析結果(n=3)Table 3 Analytical results of OH-PAHs in the urine samples of smoking volunteers (n=3)
2.5 與文獻方法對比
將本方法同文獻[3-5,14]方法進行對比(見表4),本方法靈敏度與文獻方法相當,且方法準確可靠,吸附劑用量僅需要4 mg, 3 min即可完成吸附,說明本方法簡單、快速、高效,可用于人尿液中OH-PAHs的常規分析。
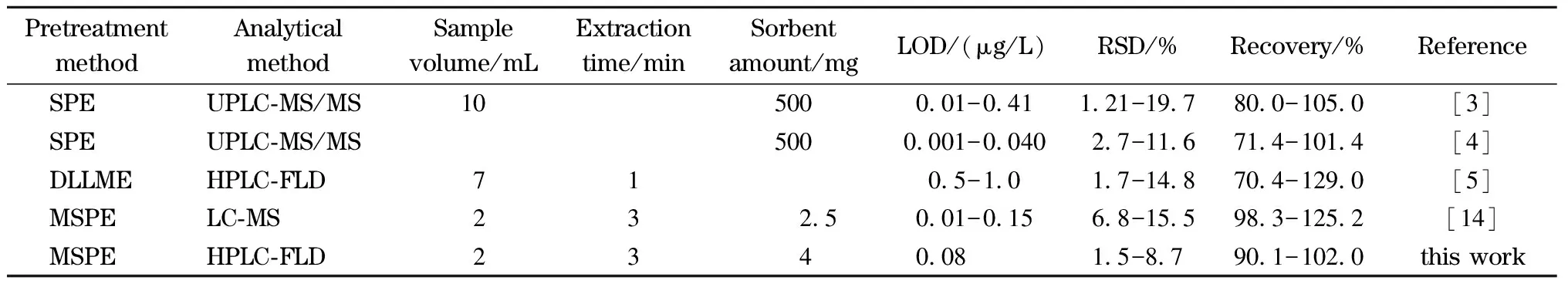
表 4 本方法與文獻方法對尿液中OH-PAHs分析結果的對比Table 4 Comparison of the analysis results of this method with methods in literatures for the determination of OH-PAHs in urine
DLLME: dispersive liquid-liquid microextraction; MSPE: magnetic solid-phase extraction.
3 結論
本研究首次以溶劑熱法制備磁性g-C3N4材料,將其用于尿液中3種OH-PAHs的前處理中,并結合HPLC-FLD對吸煙志愿者的尿液樣品進行分析。本方法能夠簡單、快速、高效的富集和凈化尿液中的3種OH-PAHs,展現了良好的應用前景。
猜你喜歡
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
甘肅教育(2020年14期)2020-09-11 07:57:42
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國衛生(2014年11期)2014-11-12 13:11:32
