轉錄因子EB調節機制的研究進展
2019-03-06 10:14:44范宜俊
中華肺部疾病雜志(電子版) 2019年1期
范宜俊 劉 偉
轉錄因子TFEB(transcription factor EB)作為MiT家族中最主要的因子之一,參與溶酶體的形成,溶酶體-自噬途徑和溶酶體胞吐的調節。TFEB在體內廣泛存在,TFEB介導的溶酶體-自噬途徑的調控在神經退行性疾病、腫瘤和代謝性疾病等多種疾病的病因和治療中都有著關鍵性的作用,因此對TFEB的調控機制的研究在各種疾病的治療中也具有展望性的意義。
一、TFEB的介紹
1. MiT家族成員的介紹: MiT家族中已有四個成員被鑒定出來:MiTF(microphthalmia-associated transcription factor, MiTF),TFEB,TFE3和TFEC。MiTF/TFE的所有成員都具有對其二聚化非常重要的高度相似的堿性螺旋-環-螺旋-亮氨酸拉鏈(basic helix-loop-helix leucine zipper, bHLH-ZiP)結構域,bHLH-ZiP結構域介導MiT家族成員形成同源或異源二聚體,進而參與靶基因轉錄的激活[1]。MiTF/TFE成員形成異二聚體的過程具有嚴格的調控,有研究發現在MiTF/TFE成員的ZiP域內存在一個保守的三殘基位移,在此引入了一個注冊外的亮氨酸拉鏈,使得MiTF/TFE成員之間只進行特定的異二聚化,同時防止與其他的bHLH-Zip域結合。然而相比異源二聚體,MiTF/TFE的同源二聚體的功能仍然未知[2]。
2. TFEB的結構特點: TFEB是由476個氨基酸殘基組成的蛋白質,主要包括富谷氨酰胺、螺旋-環-螺旋(helix-loop-helix, HLH)、亮氨酸拉鏈(leucine-zipper, LZ)和富脯氨酸等模體。TFEB能以二聚體的形式與DNA結合,通過識別啟動子 E-box、M-box啟動相應基因的轉錄,發揮其生理功能。最初TFEB被確定為MiTF/TFE家族中具有調節眾多溶酶體和自噬基因表達能力的唯一成員,廣泛表達于各類細胞,參與多種生物功能,近年來發現MiTF/TFE家族中的另一成員TFE3在應激條件下的部分組織中對溶酶體自噬也有著調節作用[3]。
二、TFEB的功能
1. TFEB對溶酶體基因表達的調控: 溶酶體是進行細胞降解和系統回收的重要組成部分,是維持細胞穩態的必要條件[4]。最初溶酶體被描述為降解細胞內廢物的靜態細胞器,但后來認為溶酶體可以通過調節基因的表達調節其生物的合成與功能[5]。Sardiello等[5]對溶酶體基因的啟動子分析得到E-box樣回文序列(GTCACGTGAC)即協調溶酶體表達和調控(coordinate lysosomal expression and regulation, CLEAR)元件的基序,顯示CLEAR元件直接與TFEB的bHLH轉錄因子結合。因此,TFEB的表達將會導致溶酶體的數量增加和效能升高,從而增強溶酶體的分解代謝活性。TFEB能誘導溶酶體的胞吐作用,Medina等[6]發現TFEB通過激活Ca2+通道蛋白MCOLN1可使Ca2+流入細胞內,促使溶酶體與質膜融合。溶酶體貯積癥時TFEB的過表達可促使細胞內的代謝廢物排至細胞外。
2. TFEB對細胞自噬的調控: TFEB與自噬基因的啟動子區結合,誘導自噬體的生物合成和自噬體-溶酶體融合以及自噬底物的降解[6]。以此為基礎,TFEB在緩解肝毒性藥物造成的肝臟損傷[7]、治療糖尿病患者的腎臟損害[8-9]和增強胃腸道腫瘤的治療效果和預后恢復等方面都起著非常重要的作用[10-11]。
TFEB過表達可促進體內脂滴的清除和線粒體損傷的降解,表明該轉錄因子在調節細胞器特異性自噬如脂肪自噬和線粒體自噬中也起作用[12]。Uchida等[13]發現在對大鼠靜脈內注射脂多糖后導致的炎癥反應中,TFEB的激活所誘導的細胞自噬在角膜細胞防御中起到了重要作用。TFEB能通過誘導溶酶體激活來調節免疫功能,TFEB激活后會抑制MHC Ⅰ類介導的外源性抗原表達,同時增強MHC Ⅱ類呈遞抗原,進而影響樹突狀細胞通過MHC途徑與T細胞結合呈遞外源性抗原的過程來影響免疫應答[14]。最近發現,在乙醇誘導的肝臟損傷模型小鼠中組織的總TFEB和核TFEB水平低于正常組,溶酶體的生物合成能力降低,而Torin-1(mTOR活性抑制劑)作用后可以提高肝組織的TFEB水平,降低乙醇對肝的損傷和脂肪變性,提示提高TFEB水平可能將對乙醇所致的酒精性脂肪肝起到保護作用[15]。
三、TFEB的調控
轉錄因子TFEB進入胞核后調控下游基因的表達,廣泛參與各種生理病理活動。其自身的核轉位也受到體內外各種蛋白分子的調節。
1. 雷帕霉素復合物1對TFEB的調控: 一般來說,正常狀態下TFEB以非活性狀態存在于胞質中[16],而在饑餓或溶酶體功能出現障礙時,TFEB迅速轉位至細胞核并激活其下游靶基因的轉錄。TFEB的細胞定位和活性狀態與其是否磷酸化有著密切的關系。TFEB蛋白中的兩個特定的絲氨酸殘基(即Ser142[17-18]和Ser211[17,19-20])在TFEB的亞細胞定位中發揮關鍵作用。當這兩個絲氨酸殘基都被磷酸化時,TFEB在胞質中保持無活性狀態[19-20],并且磷酸化的Ser211會與伴侶蛋白14-3-3結合后停留在胞質中,抑制TFEB的核轉位[20]。
哺乳動物雷帕霉素復合物1(mTORC1)和細胞外信號調節激酶2(ERK2,也稱為MAPK1)是饑餓條件下能使大多數細胞內的TFEB去磷酸化的主要蛋白激酶[20-21]。Sancak等[22-23]發現mTORC1的激活發生在溶酶體膜上。在營養物質充足的情況下,V-ATPase復合物促進溶酶體信號復合物小Rag GTP蛋白的活化,mTORC1的組件Raptor與激活的Rag GTPases結合,并被募集到溶酶體膜上,通過GTPase Rheb促進其活性。同時大量的TFEB也被活化的Rag GTP蛋白復合物募集到溶酶體膜上,通過mTORC1促進TFEB的S142、S211位點磷酸化[24]。當機體轉于饑餓狀態或應激時,溶酶體腔內的氨基酸含量降低使Rag GTPases失活,mTORC1變得無活性并隨后從溶酶體膜釋放,TFEB的S142、S211位點失去抑制并去磷酸化,與伴侶蛋白14-3-3分離。去磷酸化的TFEB進入胞核與核內CLEAR序列結合,促進下游溶酶體基因的表達,增強溶酶體的生物合成及生理功能[25],見圖1。
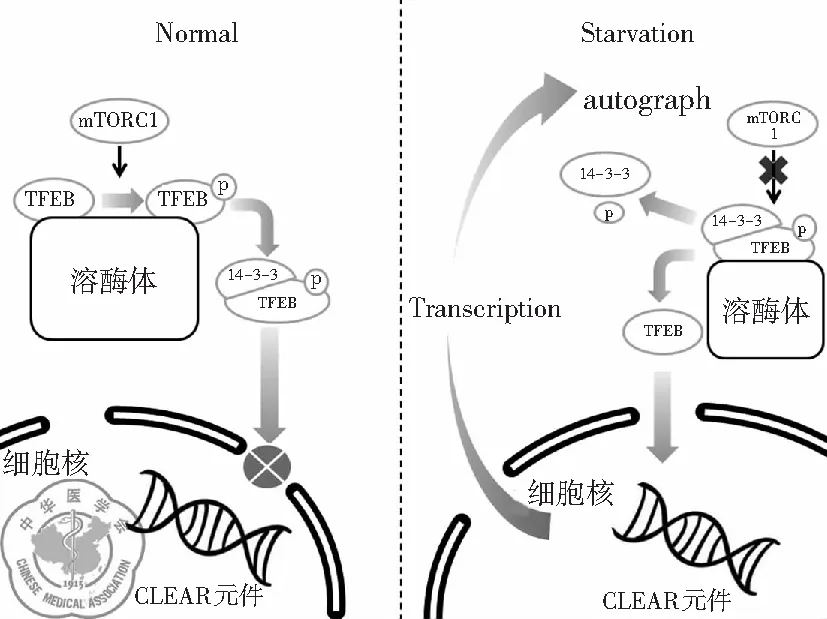
圖1 mTORC1對TFEB的調控
2. 外源性氧化劑的負反饋調節: Medina等[26]發現溶酶體基因表達的過程伴隨著溶酶體內的Ca2+而發生。變化通過Ca2+通道黏脂蛋白1(mucolipin1, MCOLN1)釋放入胞漿激活磷酸酶、鈣調磷酸酶,鈣磷酸酶又將TFEB去磷酸化并促進其向核移位。當MCOLN1減少時,則會抑制溶酶體Ca2+釋放和鈣調磷酸酶活化,從而阻止營養物剝奪時TFEB去磷酸化和自噬。這也暗示了通過調節胞漿Ca2+的含量也可以間接調控溶酶體的活性。外源性氧化劑或增加的線粒體內活性氧(reactive oxygen species, ROS)水平例如魚藤酮、過氧化氫或羰基氰化物間氯苯腙(carbonyl cyanide m-chlorophenylhydrazone, CCCP)等可特異性地激活溶酶體TRPML1通道[27],誘導溶酶體Ca2+釋放,誘導自噬和溶酶體合成[25]。因此TRPML1是位于溶酶體膜上的ROS傳感器,調節ROS水平可以協調自噬依賴性負反饋程序以減輕細胞中的氧化應激損傷和增強溶酶體的功能[25]。
3. TFEB的絲氨酸殘基位點改變帶來的影響: 最近的研究還表明,TFEB的另一個臨界位點Ser122對于通過mTORC1調節的TFEB亞細胞定位非常重要。具體地講,經抑制mTORC1后的TFEB核定位被TFEB的Ser122突變阻斷,這種突變也抑制了Torin1所誘導的溶酶體生物合成。這揭示了mTORC1調節TFEB的新機制,表明mTORC1對溶酶體生物發生至關重要[21]。
上述信號級聯放大反應強調了溶酶體作為信號樞紐的核心作用,該信號中樞能夠感知營養物的可用性并協調轉錄程序的激活,從而允許細胞不斷調整以適應逐步增長的代謝需求。許多可以調節TFEB活性的因子,如大多數v-ATPase亞基,Ca2+通道MCOLN1都位于溶酶體上,這表明溶酶體適應環境變化是一種受多個反饋回路共同調節的自我維持反應。
4. 細胞外信號調節激酶2對TFEB的調控: 同樣的,在細胞營養充足的情況下,絲裂原活化蛋白激酶1(mitogen-activated protein kinase 1, MAPK1)家族的細胞外信號調節激酶2(extracellular signal regulated kinase 2, ERK2)介導TFEB的ser142磷酸化,使TFEB定位在胞質內。而在饑餓狀態下時,ERK2與TFEB解離,TFEB去磷酸化后進入胞核中,促進微管相關蛋白1A/1B輕鏈3B(microtubule-associated proteins 1A/1B light chain 3B, MAPLC3B)、液泡分揀蛋白11(vacuolar protein sorting 11, VPS11)的合成和自噬相關基因9B(autophagy-related gene 9B, ATG9B)的表達[28]。
5. 蛋白激酶對TFEB的調節: 在營養充足的狀態下,細胞可通過介導自身內外界信號蛋白激酶C信號通路(PKC signaling pathway)促進MiTF/TFE的表達、溶酶體的合成和自噬底物的降解[29]。巨大戟烷型二萜類化合物中的HEP14和HEP15可以激活蛋白激酶C(protein kinase C, PKC)家族的成員PKCα和PKCδ,導致糖原合成激酶3β(glycogen synthase kinase-3β, GSK3β)失活,使得GSK3β對TFEB上第134和138位的絲氨酸位點的磷酸化作用解除,TFEB去磷酸化激活入核,促進溶酶體的生成。除了通過解除對TFEB的磷酸化來增強自噬外,被HEP激活后的PKCα可以通過激活胞內的c-Jun氨基末端激酶(c-Jun N-terminal kinase, JNK)和p38絲裂原活化蛋白激酶(p38 mitogen-activated protein kinase, p38MAPK)使溶酶體相關的抑制因子含KRAB和SCAN3結構域的鋅指蛋白(zine finger protein with KRAB and SCAN domains, ZKSCAN3)失去活性并移位至細胞質中,解除ZKSCAN3對溶酶體合成的抑制作用[30-31]。用核因子κB配體受體激活劑(RANKL)刺激骨細胞后,TFEB的C末端區域的三個絲氨酸殘基被蛋白激酶Cβ(PKCβ)磷酸化,保證了溶酶體生物合成的穩定性[32]。
蛋白激酶B(Akt)磷酸化TFEB Ser467位點并以獨立于mTORC1的機制抑制TFEB核轉位。自噬增強劑海藻糖通過減少Akt活性來激活TFEB。Batten病是一種神經元內的溶酶體產生的蠟樣脂褐質存積的神經變性疾病,將海藻糖給予患Batten病的小鼠模型可增強蛋白脂質聚集體的清除,可以減少神經病理的發生并延長患病小鼠的存活[33]。Akt的藥理作用可促進患有各種溶酶體疾病患者細胞中蛋白脂質體的清除,并提示該方法的廣泛適用性。這些發現為TFEB介導的神經退行性儲存疾病中細胞蛋白脂質體的清除開辟了新的思路[34]。此外,對于TFEB旁系同源物,MiTF和TFE3也觀察到有類似的結果[3]。
6. 姜黃素的衍生物C1對TFEB的調節: 合成姜黃素的衍生物C1被鑒定為TFEB的新型活化劑。化合物C1與TFEB的N末端特異性結合并促進TFEB核轉位,證實化合物C1在體內外增強自噬和溶酶體生物合成。并且化合物C1是TFEB的有效口服活化劑,是用于治療神經變性疾病的潛在治療劑[35]。隨后發現槲皮素也有著類似的作用,槲皮素能夠逆轉乙醇對TFEB核轉位的抑制作用,并表現出與Torin 1相似的作用,可促進TFEB的核轉位,改善由乙醇導致的溶酶體自噬功能障礙[36]。
自噬作為細胞穩態的重要功能,廣泛參與了各種生理病理過程。MiTF/TFE轉錄因子通過對溶酶體-自噬途徑中相關基因的調節,成為細胞內能量調控和細胞清除的主要調節因子。正常細胞通過對MiTF/TFE的調控維持自身的穩態和營養平衡,但是進一步機制的研究仍需要探清各種內外界刺激和多種抑制信號如何作用于不同類型的細胞而產生不同的應激反應。TFEB介導的溶酶體-自噬的調控與多個系統和功能相關,對細胞內TFEB調節機制的研究,不僅有助于闡述局部炎癥反應、免疫調節功能和神經系統性疾病可能的分子機制,也為以后對人體各種疾病的臨床治療和預防提供了一個新的方向和理論依據。
