基于miRNA-mRNA調控網絡的幽門螺桿菌感染相關病變分子機制研究
2019-03-01 06:42:56班春梅鄧婭婕
胃腸病學 2019年1期
付 偉 謝 東 班春梅 曹 潔 王 松 鄧婭婕
中國人民解放軍聯勤保障部隊第925醫院消化血液科(550009)
背景:幽門螺桿菌(Hp)感染與慢性胃炎、十二指腸潰瘍、胃癌以及多種胃腸外疾病相關,然而其分子致病機制及其與宿主間的相互作用尚不明確。目的:基于miRNA-mRNA調控網絡進行生物信息學分析,為闡明Hp與宿主間的相互作用以及Hp感染可能造成的病變提供線索。方法:使用GEO數據集篩選Hp感染相關miRNAs和mRNAs,檢索差異表達miRNAs的靶基因并與差異表達mRNAs取交集以構建miRNA-mRNA調控網絡,對靶基因進行GO和KEGG富集分析,構建蛋白質相互作用網絡,分析Hp感染與疾病的相關性。結果:靶基因功能富集分析顯示富集最顯著的基因集為趨化因子受體結合趨化因子。蛋白質相互作用網絡中共有28個核心基因,疾病連接性分析顯示Hp感染與癌癥以及消化、神經、心血管、呼吸、免疫系統疾病相關。結論:本研究提供了一種基于miRNA-mRNA調控網絡預測病原體與人類疾病相關性的方法,并發現Hp感染可增加多種胃腸外疾病風險。
幽門螺桿菌(Helicobacterpylori, Hp)是一種革蘭陰性致病菌,長期攜帶Hp可顯著增加慢性胃炎、十二指腸潰瘍、胃腺癌和胃黏膜相關淋巴樣組織淋巴瘤的發生風險,并與多種胃腸外疾病相關[1]。然而Hp感染的分子致病機制及其與宿主間的相互作用迄今尚未完全明確。
基因轉錄失調在人類疾病中起有重要作用,而微RNA(microRNAs, miRNAs)通過在轉錄后水平下調靶基因表達,在諸多生理和病理過程中發揮重要調控作用[2]。因此Hp感染相關miRNAs及其靶基因的鑒定可能為相關分子致病機制的認識提供新的見解。高通量基因表達數據庫Gene Expression Omnibus(GEO; http://www.ncbi.nlm.nih.gov/geo)是一個可以儲存和發布高通量微陣列以及其他形式功能基因組數據的公共存儲庫[3]。本研究通過從GEO數據集中鑒定Hp感染相關miRNAs及其靶mRNAs,構建miRNA-mRNA調控網絡并進行生物信息學分析,以期為闡明Hp與宿主之間的相互作用以及Hp感染可能造成的病變提供線索。
材料與方法
一、GEO數據集
本研究使用的GEO數據集為GSE19769和GSE5081,前者為10例Hp陰性和9例Hp陽性人胃黏膜標本的miRNAs微陣列數據,后者為16例Hp陰性和16例Hp陽性糜爛胃黏膜與鄰近正常胃黏膜標本的全基因組寡核苷酸微陣列數據。
二、方法
1. 篩選差異表達miRNAs和mRNAs:采用GEO2R對所有表達數據進行分析[4]。調整P值默認使用Benjamini和Hochberg(錯誤發現率)方法以降低假陽性率。MiRNAs數據集cut-off值設定為調整后P<0.05且|log10FC|>2;mRNAs數據集cut-off值設定為調整后P<0.05且|log10FC|>1.5。使用MeV 4.9通用微陣列分析工具進行差異表達的分層聚類并可視化。
2. MiRNA-mRNA調控網絡構建:從DIANA-TarBase 8.0中檢索差異表達miRNAs的靶基因[5],并與mRNAs數據集中的差異表達基因取交集。使用Cytoscape 3.6.1生物信息分析軟件產生miRNA-mRNA調控網絡[6]。
3. GO(Gene Ontology)、KEGG(Kyoto Encyclopedia Genes and Genomes)富集分析和蛋白質相互作用網絡構建:使用Metascape對Hp感染相關miRNAs靶基因進行GO功能和KEGG信號通路富集分析。使用STRING 10.0構建蛋白質相互作用網絡,cut-off值設定為置信評分≥0.9[7],Cytoscape 3.6.1進行可視化,CentiScaPe 2.2進行生物網絡分析,計算節點屬性,degree>20閾值鑒定為核心基因[8]。
4. Hp感染與疾病相關性分析:使用比較毒理基因組數據庫(Comparative Toxicogenomics Database)“set analyzer”工具鑒定與Hp感染相關基因相關的疾病[9],cut-off值設定為Bonferroni校正P<0.05,P值越小,基因與疾病之間的連接越可靠。
結 果
一、Hp感染相關miRNAs和mRNAs
對GEO數據集GSE19769、GSE5081的分析分別鑒定出42個差異表達miRNAs和633個差異表達mRNAs(圖1)。
二、MiRNA-mRNA調控網絡
42個Hp感染相關miRNAs從TarBase數據庫中共獲得10 617個靶基因,經與Hp感染相關mRNAs交集,最終獲得194個靶基因。MiR-153-5p靶基因最多,共52個,miR-95-5p、miR-577、miR-548b-5p、miR-519e-5p亦均調控10個以上靶基因。MiRNAs對靶基因的調控為下調其表達,根據基因表達上調或下調建立關系圖(圖2),圖中122個靶基因可能每列包含來自特定基因的數據,每行包含來自單個樣本的數據;紅色為上調,綠色為下調在Hp定植和持續感染中發揮重要作用。
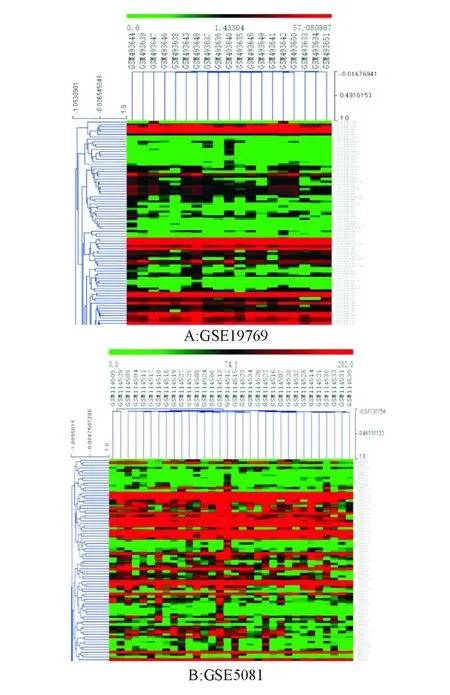
圖1 Hp感染相關miRNAs(A)和mRNAs(B)的鑒定(微陣列分析熱圖)
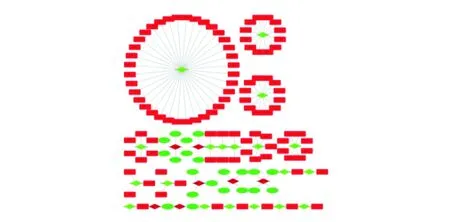
長方形和橢圓形為基因,菱形為miRNAs(紅色為上調,綠色為下調);與miR-153-5p(圖中左上方環形中心)相關性最強的36個靶基因為WWC1、UTP23、UQCC1、TTL、TRPM3、TRDN、SPAG16、SLC8A1、SFSWAP、SAMD12、RAB39B、PPP1R16B、PMP2、NIN、NETO1、NALCN、MYEF2、MSR1、MS4A1、MC2R、LPCAT1、KLHL6、KLF12、KIAA0408、KCTD16、KCNN3、KCND3、HMGA2、GPNMB、FCRL4、FAM65B、EPHA3、ELOVL2、CYP7A1、CDH13、ALDOB
三、MiRNA靶基因功能富集分析
對Hp感染相關miRNAs靶基因進行GO-BP、KEGG途徑富集分析,富集最顯著的基因集為趨化因子受體結合趨化因子(R-HSA-380108)和金屬離子內穩態(GO:0055065)(圖3A)。此外,分析還顯示Hp感染與免疫應答調節信號通路(GO:0002764)、腎單位發育(GO:0072006)等有關,各基因集之間呈現復雜的網絡關系(圖3B)。
四、Hp感染與疾病的相關性
圖1中Hp感染相關miRNAs靶基因的蛋白質相互作用網絡見圖4。28個核心基因的疾病連接性分析顯示Hp感染與包括癌癥以及消化系統、神經系統、心血管、呼吸道和免疫系統疾病在內的48種人類疾病相關。
討 論
本研究利用GEO數據集GSE19769、GSE5081篩選Hp感染相關miRNAs和mRNAs,并基于Tarbase數據庫構建miRNA-mRNA調控網絡,結果顯示miR-153-5p、miR-95-5p、miR-577、miR-548b-5p、miR-519e-5p為中心miRNAs,其中miR-153-5p靶基因最多,圖2網絡中列出了與之相關性最強的36個靶基因。MiR-153-5p、miR-95-5p、miR-577等原則上可作為診斷性生物學標志物和治療靶點。既往研究發現,胃
癌組織中miR-153表達下調,其靶基因Snail表達上調,從而介導腫瘤侵襲、轉移[10];此外,miR-153在胃癌中還參與了超保守區域轉錄子Uc.416+A的調節,進而可調節腫瘤細胞生長[11]。Hsa-miR-95在胃印戒細胞癌中表達下調,在腸型胃腺癌中表達上調,可能與印戒細胞癌的高侵襲轉移性和化療耐藥密切相關[12]。而受這些miRNAs調控的靶基因在Hp感染和相關疾病發生、發展進程中可能亦發揮重要生物學作用。靶基因功能富集分析顯示富集最顯著的基因集為趨化因子受體結合趨化因子和金屬離子內穩態,而一些先前未被重視的生物過程和途徑亦可能與Hp感染有關。
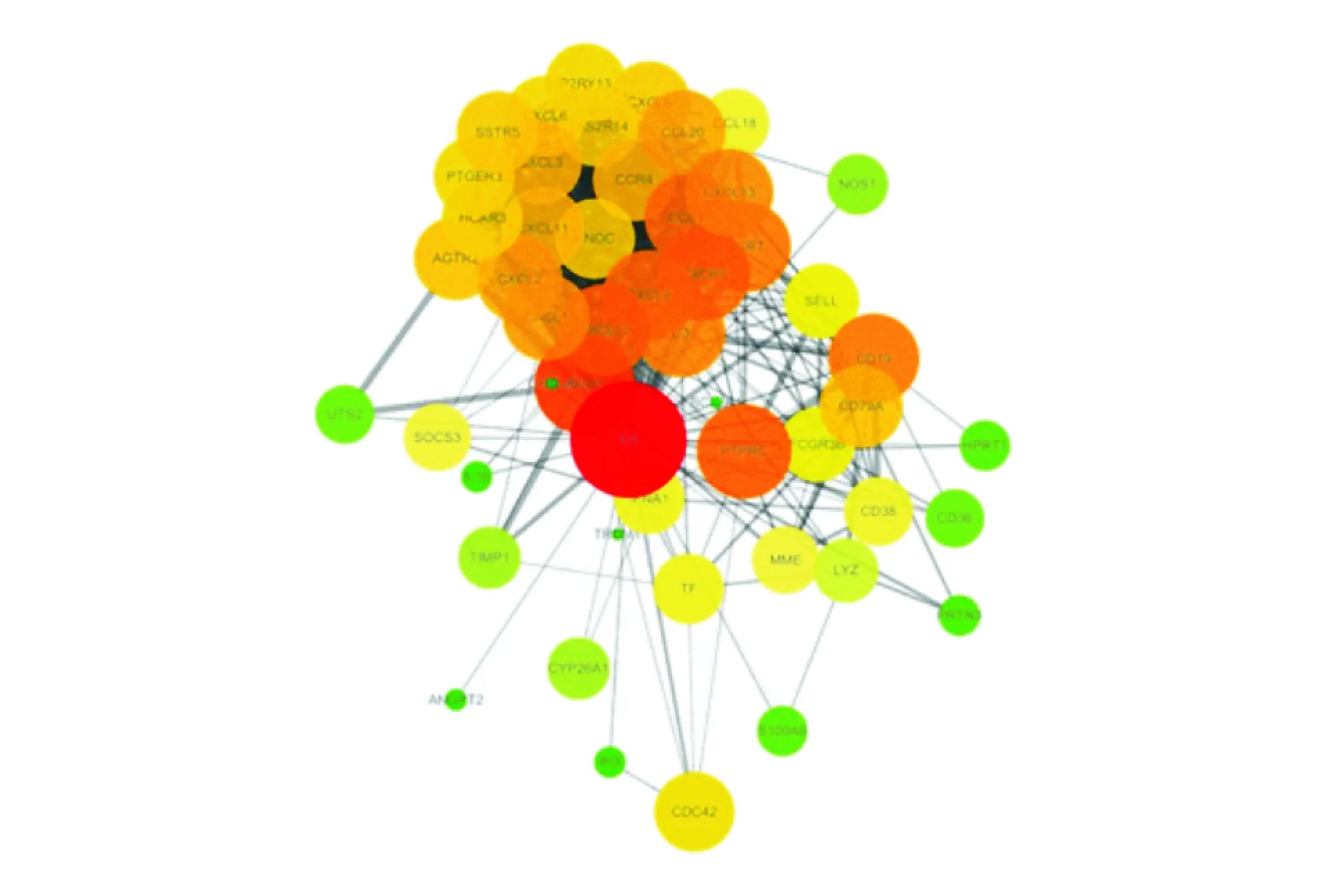
根據degree閾值鑒定得到28個核心基因(IL8、KNG1、PTPRC、CXCL10、CXCR5、CCL19、CCR7、CD19、CXCL9、C3、CXCL13、CXCL1、CCL20、CXCL2、AGTR2、CCR4、CD79A、CXCL11、CXCL3、CXCL5、SSTR5、CXCL6、HCAR3、P2RY13、PNOC、PTGER3、TAS2R14、CDC42),圖形體積越大,degree閾值越高
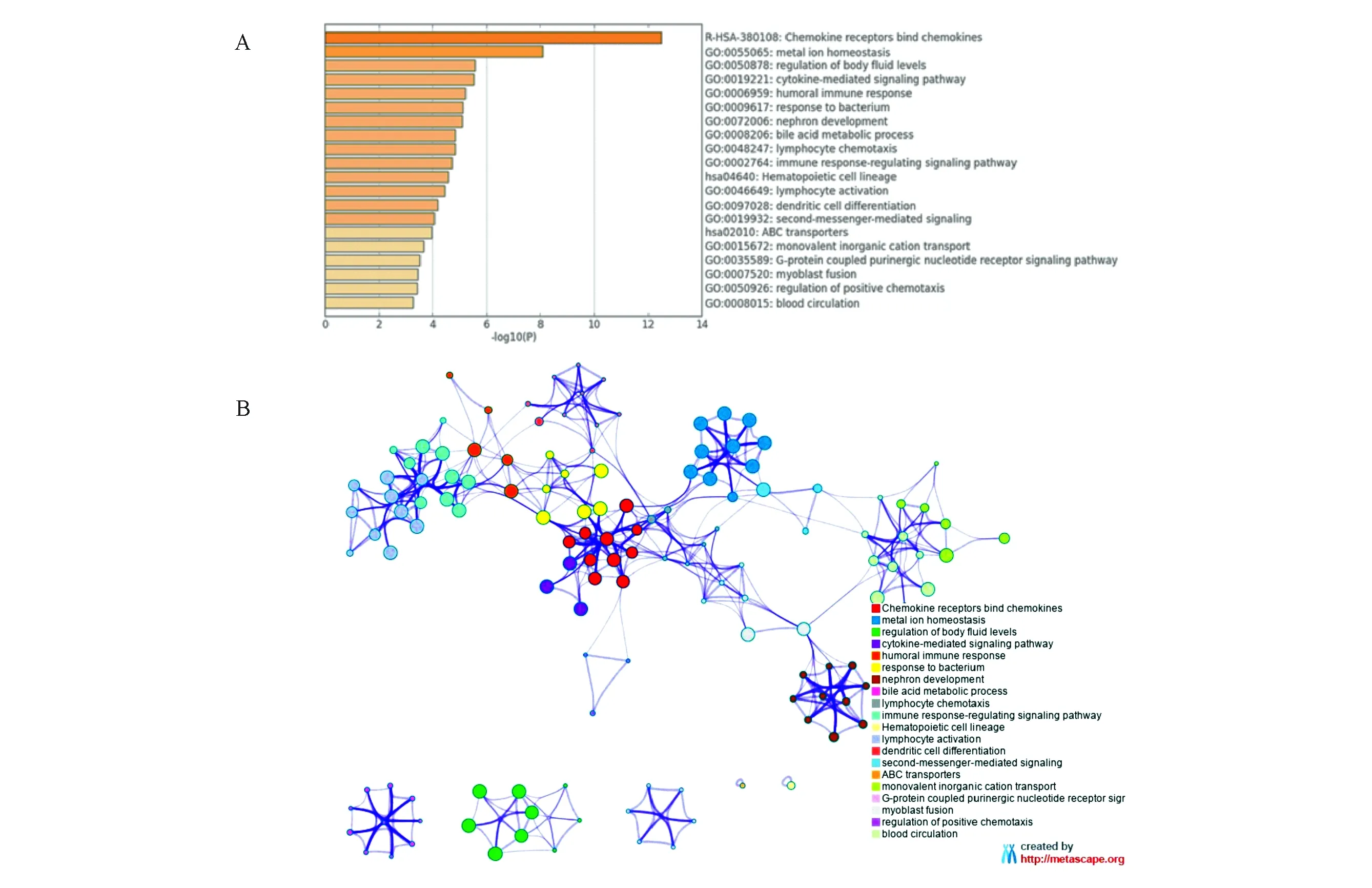
A:Hp感染相關miRNAs靶基因Metascape途徑富集分析的前20個簇,條形長度為每個聚類中最佳評分項的log10(P);B:Metascape分析前20個聚類富集項之間的關系(相同顏色的節點屬于同一個群集)
共28個基因被確定為Hp感染相關miRNAs靶基因蛋白質相互作用網絡的核心基因,包括白細胞介素-8(IL-8)以及一系列趨化因子受體和配體。IL-8作為一種炎癥細胞因子,可由Hp感染誘導表達,并與胃癌進展和轉移密切相關[13]。人胃上皮細胞定植的Hp經Toll樣受體2(TLR2)識別,可誘導CXCL1、CXCL5、CXCL8等趨化因子表達[14]。仍有一些核心基因如KNG1的功能尚未確定,為未來Hp感染的研究提供了新的方向。
對上述28個核心基因的疾病連接性分析顯示Hp感染與癌癥以及消化、神經、心血管、呼吸、免疫系統疾病相關,與既往研究結果相符[15-18]。當前證據支持微生物失調在人類疾病中發揮重要的信使作用,Hp感染可能的致病機制包括分子模擬、慢性炎癥、損傷DNA等,其不僅直接損傷宿主細胞,而且可損傷黏膜屏障、改變消化道微生物群。
綜上所述,本研究嘗試了通過構建miRNA-mRNA調控網絡預測病原體與人類疾病相關性的新方法,并發現Hp感染可增加多種胃腸外疾病風險,相關核心基因的鑒定可能為未來研究提供方向。
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中國特種設備安全(2018年11期)2019-01-08 02:08:32
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
山東工業技術(2016年15期)2016-12-01 05:31:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:46
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
山東女子學院學報(2014年6期)2014-03-01 02:24:55
