高效液相色譜-串聯質譜法測定蜂蜜中9種農藥殘留
2019-01-05 01:56:16朱之烔寧倩倩沈偉健陳惠蘭沈崇鈺
色譜 2019年1期
朱之烔, 柳 菡*, 寧倩倩, 張 健, 沈偉健,陳惠蘭, 沈崇鈺, 謝 文
(1. 南京海關動植物與食品檢測中心, 江蘇 南京 210019; 2. 杭州海關, 浙江 杭州 310016)
苯并咪唑類農藥是一類低毒高效內吸性的廣譜殺菌劑[1]。其中多菌靈(carbendazim)、甲基托布津(thiophanate-methyl)及乙基托布津(thiophanate-ethyl)較為常見,在水果蔬菜及農副產品中檢出率較高[2,3]。其中多菌靈的急性毒性比較低,但它在自然界中降解半衰期長,可通過食物鏈蓄積對人類身體健康尤其是生殖系統造成危害[4]。而甲基托布津不穩定,會在植物體內中轉化為多菌靈[5]。洛苯達唑(lobendazole,即ethylN-(1H-benzimidazol-2-yl)carbamate,簡稱EBC)是乙基托布津在生物體內的代謝物[5]。研究表明,其小鼠靜脈注射半數致死量(lethal median dose, LD50)為180 mg/kg,具有一定的生殖毒性,能夠影響胎兒的肌肉骨骼發育,具有一定的致畸性[6]。
新煙堿類農藥是一類昆蟲乙酰膽堿受體激動劑,能引發劇烈的神經毒害效應[1]。其中常見的有吡蟲啉、啶蟲脒、噻蟲胺、噻蟲嗪等。雖然對哺乳動物無毒害作用,但新煙堿類殺蟲劑會對蜜蜂免疫系統和中樞神經系統等造成損傷,甚至引起蜜蜂急性中毒死亡[7],不僅僅使養蜂業承受了巨大的經濟損失,更會危害整個生態系統。
蜂蜜是一種營養豐富的純天然滋養食品。但由于蜜蜂在采蜜過程中采集了被農藥污染的花粉及蜂農不恰當使用的農藥,導致蜂蜜中的農藥殘留量有可能超標。研究表明,全球超過75%的蜂蜜含有至少一種農藥的殘留[8]。目前,歐盟規定蜂蜜中多菌靈和苯菌靈總和不超過1.0 mg/kg[9],并自2013年以來限用并將全面禁用3種新煙堿類殺蟲劑(吡蟲啉、噻蟲胺和噻蟲嗪)[10]。而我國暫未規定蜂蜜中這兩類農藥的殘留限量。
目前國內分別檢測苯并咪唑類和新煙堿類農藥殘留的文獻有很多,包括了紫外可見分光光度法[11]、免疫化學法[12,13]、高效液相色譜法[14,15]、高效液相色譜-質譜聯用法[16,17]和離子交換色譜法[18]等方法。其中,高效液相色譜-質譜聯用法抗干擾能力強,靈敏度高,是主要的分析方法。但針對蜂蜜基質的研究不多,且同時涉及兩類農藥的較少,對洛苯達唑的研究報道極少。蜂蜜中含有大量的糖類物質和少量的有機雜質,蜂蜜中農藥殘留的前處理方法有液液萃取法[16]、分散固相萃取法[17]和固相萃取法[19]等。由于基質效應的存在,報道的研究大多使用基質校準曲線。本研究針對4種苯并咪唑類農藥(多菌靈、甲基托布津、乙基托布津、洛苯達唑)和5種新煙堿類農藥(吡蟲啉、啶蟲脒、呋蟲胺、烯啶酰胺和氟啶蟲酰胺),采用同位素內標法定量,在此基礎上比較了不同的溶解試劑和前處理凈化方式,建立了蜂蜜中9種苯并咪唑類和新煙堿類農藥殘留的全自動固相萃取-高效液相色譜-串聯質譜測定方法。
1 實驗部分
1.1 儀器與試劑
Agilent 1260 Infinity II高效液相色譜儀和Agilent 6470A三重四極桿質譜儀(美國Agilent公司);真空氮氣吹干儀(美國Caliper公司); Heraeus Mutifuge X1R離心機(美國Thermo-Fisher公司); Smart-N超純水機(美國Millipore公司);親水親脂平衡(hydrophilic-lipophilic balance, HLB)固相萃取柱(美國WATERS公司); Preval SPE 304+全自動固相萃取裝置(北京普立泰科儀器公司)。
多菌靈、甲基托布津、乙基托布津、吡蟲啉、啶蟲脒、呋蟲胺、烯啶蟲胺和氟啶蟲酰胺,純度均高于97%(德國Dr. Ehrenstorfer公司);洛苯達唑,純度高于99%(北京曼哈格生物科技有限公司);多菌靈-D4、吡蟲啉-D4,純度均高于97%(美國Sigma-Aldrich公司);甲醇、甲酸、乙腈、乙酸乙酯,均為色譜純(德國Merck公司);水合磷酸二氫鉀,分析純(南京化學試劑有限公司);N-丙基乙二胺(PSA), 40~60 μm(美國Sepax Technologies公司);實驗用蜂蜜樣品均購自超市。
1.2 標準溶液的配制
標準儲備液:準確稱取9種農藥標準品各5.0 mg,分別用甲醇溶解并定容至50 mL,配制成質量濃度為100 mg/L的標準儲備液(多菌靈和洛苯達唑可以加入適量甲酸助溶),放置在-18 ℃的冰箱中冷凍保存。
混合標準工作溶液:分別取適量上述9種農藥的標準儲備液,用甲醇逐級稀釋,配制成1 mg/L和0.1 mg/L的混合標準工作液,放置在-4 ℃的冰箱中保存。
內標儲備液:準確稱取10.0 mg的多菌靈-D4、吡蟲啉-D4,用甲醇溶解并定容至100 mL,配制成100 mg/L的內標儲備液,放置在-18 ℃的冰箱中冷凍保存。
內標工作溶液:取適量內標儲備液,用甲醇稀釋,配制成0.1 mg/L內標工作溶液,放置在-4 ℃的冰箱中保存。
標準曲線的配制:準確吸取內標工作液100 μL、標準工作液適量,以甲醇-水溶液(3∶7, v/v)配制質量濃度為0.002、0.005、0.01、0.02、0.05 mg/L的溶液,現配現用。
磷酸鹽緩沖液的配制:準確稱量27.2 g水合磷酸二氫鉀固體,先用800 mL水溶解,再用6 mol/L NaOH溶液調節pH至7.8,加水至1 000 mL。
1.3 樣品前處理
1.3.1提取
準確稱量提前混勻的蜂蜜樣品(1.00±0.05) g,置于50 mL塑料離心管中,加入100 μL內標溶液、15 mL磷酸鹽緩沖溶液,加蓋渦旋使溶液充分溶解、混勻;超聲15 min;用濾紙過濾至20 mL玻璃試管中,待凈化。
1.3.2凈化
先將上述玻璃試管放入全自動固相萃取儀中,再將HLB固相萃取柱放入全自動固相萃取儀中,預先設計好程序(HLB固相萃取柱用5 mL甲醇活化,5 mL水平衡,采用3 mL/min的流速上樣,5 mL水淋洗,5 mL甲醇洗脫),樣品按此程序進行凈化;洗脫液在45 ℃水浴下氮氣吹干;用甲醇-水溶液(3∶7, v/v)定容至1.0 mL,渦旋30 s,過0.22 μm濾膜至進樣小瓶中,供液相色譜-質譜聯用儀測定。
1.4 分析條件
1.4.1色譜條件
色譜柱:SVEA C18色譜柱(100 mm×2.1 mm, 5 μm);柱溫:40 ℃;流動相A相為0.1%(v/v)甲酸水溶液,B相為0.1%(v/v)甲酸甲醇溶液;流速:0.4 mL/min。梯度洗脫程序:0~1 min, 95%A; 1~2 min, 95%A~40%A; 2~5 min, 40%A~0%A; 5~6 min, 0%A~95%A。進樣量:10 μL。
1.4.2質譜條件
離子源:電噴霧離子(ESI)源,正離子模式;多反應監測(MRM)模式;噴霧電壓:4 000 V;干燥氣溫度:350 ℃;干燥氣流速:10 L/min;霧化氣壓力:344 kPa;鞘氣溫度:400 ℃;鞘氣流速:12 L/min; 11種化合物的其他質譜參數見表1。
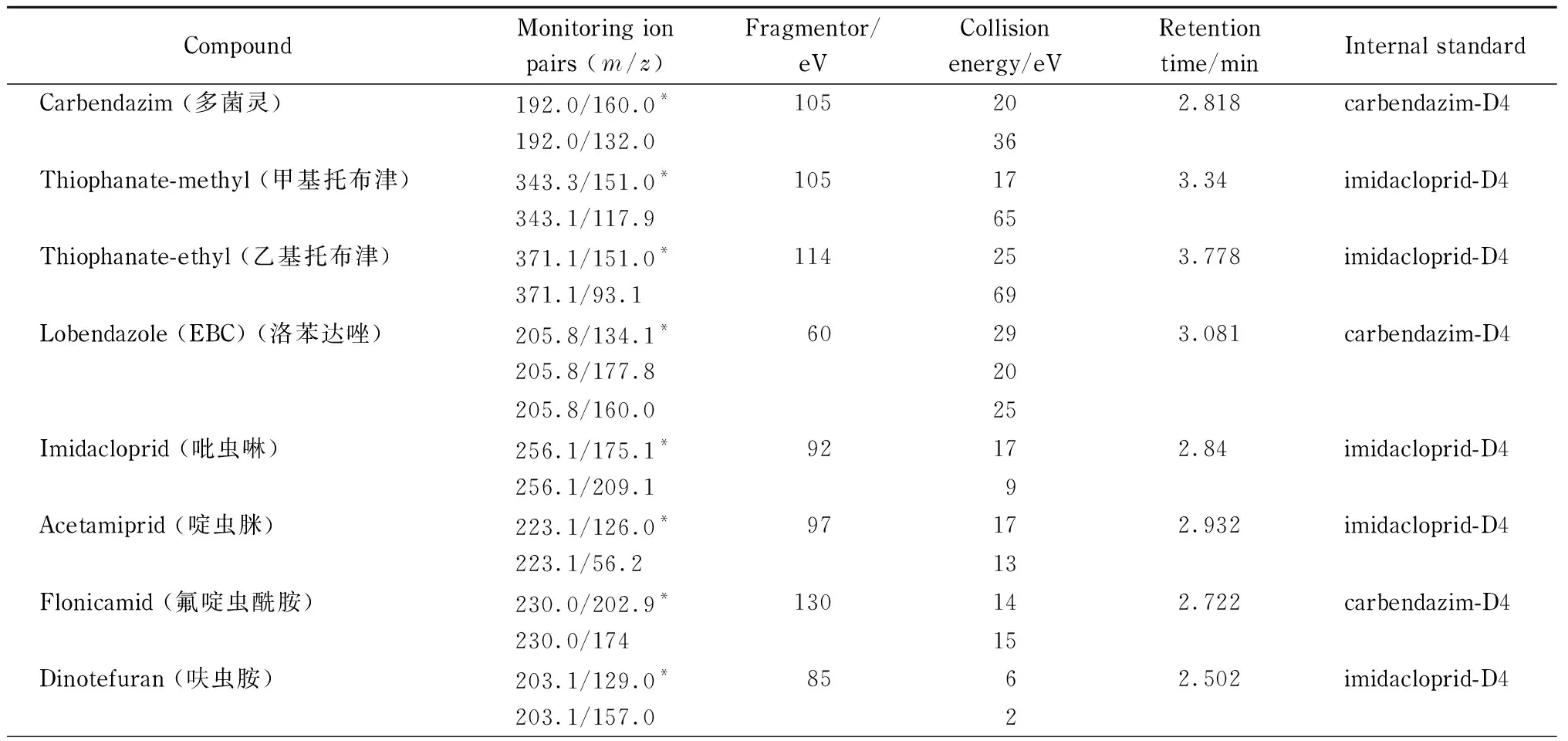
表 1 11種化合物的監測離子對、毛細管出口電壓、碰撞能量、保留時間及內標物Table 1 Monitoring ion pairs, fragmentor, collision energy, retention time and internal standards of the 11 compounds

表 1 (續)Table 1 (Continued)
* Quantitative ion.
2 結果與討論
2.1 前處理方法的優化
蜂蜜中含有大量的糖類物質和少量的有機雜質,本實驗著重于前處理方法的研究。在50.0 μg/kg水平下,各前處理方法分別做3個平行的陰性樣品加標試驗,以平均回收率判定方法的優劣。

圖 1 不同溶劑對乙酸乙酯提取后的目標化合物回收率的影響Fig. 1 Effect of the different dissolving reagents on the recoveries of the target compounds after extraction with ethyl acetate
2.1.1樣品溶解試劑的確定
由于蜂蜜基質的特殊性,直接加入有機試劑時樣品易變成膠質狀,故先用少量水溶解、稀釋蜂蜜,使其成為均相體系。研究最初采用液液萃取法進行預處理,而乙酸乙酯是常用的提取試劑,且據報道[16]對多菌靈等農藥有較高的回收率。故在樣品內加入乙酸乙酯,渦旋、超聲、離心后,將提取液氮吹濃縮至干,復溶后進行液相色譜-質譜聯用分析。實驗結果如圖1所示,僅有多菌靈、洛苯達唑、吡蟲啉和啶蟲脒的回收率達到80%,呋蟲胺約40%,而甲基托布津、乙基托布津、烯啶蟲胺和氟啶蟲酰胺的回收率不足25%。
蜂蜜溶于水后的溶液偏酸性,而且研究中的農藥呈中性至弱堿性,可能會以離子形式存在于蜂蜜水溶液中,不利于有機溶劑提取。因此,考慮使用偏堿性的磷酸鹽緩沖液(pH=7.8)來溶解和稀釋蜂蜜,使目標物在此pH值下以分子形式存在,再加入乙酸乙酯超聲提取,提取液濃縮定容后,進行液相色譜-質譜聯用分析,結果如圖1所示。在緩沖液溶解樣品后,呋蟲胺的回收率提高明顯,達到了70%以上。甲基托布津、乙基托布津、烯啶蟲酰胺和氟啶蟲酰胺也均有一定提高,但仍不足50%。多菌靈、洛苯達唑、吡蟲啉和啶蟲脒也均有較好的回收率。故選用磷酸鹽緩沖液作為蜂蜜樣品的溶解試劑。
2.1.2樣品前處理方法的選擇
研究比較了乙酸乙酯萃取法、QuEChERS法和固相萃取法的凈化效果。QuEChERS法是以乙腈作提取溶劑,加入適量的鹽,以鹽析作用促進提取,分散固相萃取法凈化來完成[20]。將樣品用少量緩沖液溶解后,加入適量乙腈超聲提取。考慮到PSA可以吸附基質中各種極性有機酸、糖類和脂肪等雜質,故在鹽析過程中加入少量PSA除雜。
固相萃取技術能夠選擇性吸附、保留目標物,除去雜質的同時,還能起到濃縮的作用。其中的HLB固相萃取柱常用于凈化蜂蜜中大量糖類及極性極強的雜質[19]。蜂蜜樣品經緩沖液溶解后,需過濾以避免固相萃取柱堵塞。過濾液經固相萃取柱凈化后,收集的洗脫液經氮吹復溶后進行液相色譜-質譜聯用分析。
由圖2可知,對于多菌靈、洛苯達唑和吡蟲啉,3種方法均有較好的回收率;對于啶蟲脒、呋蟲胺,乙酸乙酯萃取法和固相萃取法的回收率可達75%以上;對于烯啶蟲胺、氟啶蟲酰胺,QuEChERS法和固相萃取法的回收率達到75%以上;對于甲基托布津和乙基托布津,僅有固相萃取法的回收率較好。故選擇HLB固相萃取法對樣品進行凈化。

圖 2 不同前處理方法對目標化合物回收率的影響Fig. 2 Effect of different pretreatment methods on the recoveries of the target compounds Method 1: extraction by ethyl acetate; method 2: extraction by QuEChERS; method 3: extraction by solid-phase extraction with hydrophilic-lipophilic balance (HLB) cartridges.
2.2 儀器條件優化
2.2.1色譜條件的優化
實驗之初以水溶液作為水相,發現甲基托布津和乙基托布津的峰形拖尾嚴重,其他化合物的峰形也存在一定程度的不對稱。而以0.1%(v/v)甲酸作水相時,各化合物的峰形對稱,如圖3所示。因此采用0.1%(v/v)甲酸溶液作水相,0.1%(v/v)甲酸甲醇為有機相。
2.2.2質譜條件的確定
研究的9種農藥以及同位素內標的分子結構均含有氨基,易形成正離子,故采用正離子模式進行電離。在MRM模式下對11種化合物的母離子、毛細管出口電壓,子離子、碰撞能量等條件進行優化,結果如表1所示。由于流動相中添加少量甲酸,11種化合物的母離子峰均為加氫峰。

圖 3 0.05 mg/L標準溶液中11種化合物的MRM色譜圖Fig. 3 MRM chromatograms of the 11 compounds in 0.05 mg/L standard solution
2.3 方法學驗證
2.3.1線性范圍、檢出限和定量限
確定實驗方法后,按照1.2節配制標準曲線,進行標準溶液的測定。以質量濃度(X, mg/L)為橫坐標,標準物的峰面積與內標物的峰面積之比(Y)為縱坐標,繪制標準曲線。結果顯示,在0.002~0.05 mg/L范圍內,9種農藥均呈現出良好的線性關系(相關系數r2≥0.99),如表2所示。在陰性蜂蜜中加標測定,3倍信噪比對應的加標水平為方法的檢出限,10倍為定量限,結果如表2所示。
2.3.2回收率和精密度
選取陰性蜂蜜樣品,分別在5.0、10.0、20.0 μg/kg 3個水平下進行加標試驗,測得的回收率和精密度如表3所示。結果顯示,蜂蜜中9種農藥的平均回收率在78.2%~104.6%之間,相對標準偏差在1.3%~14.3%之間。

表 2 9種農藥的回歸方程、相關系數、檢出限和定量限Table 2 Regression equations, correlation coefficients (r2), LODs and LOQs of the nine pesticides
Y: peak area ratio of the quantitative ion of pesticide to internal standard;X: mass concentration, mg/L.

表 3 陰性蜂蜜中9種農藥的加標回收率和相對標準偏差(n=6)Table 3 Recoveries and RSDs of the nine pesticides in blank honey (n=6)
2.4 實際樣品檢測
用上述建立的方法對實際蜂蜜樣品進行檢測。選擇30批蜂蜜樣品進行檢測,多菌靈、吡蟲啉和啶蟲脒分別有不同程度的檢出。其中多菌靈檢出17批,殘留量在5~10 μg/kg之間。吡蟲啉和啶蟲脒分別檢出13批和12批,檢出量分別為5~20 μg/kg和5~10 μg/kg。其余農藥未檢出。
3 結論
本研究建立了蜂蜜樣品經緩沖液溶解、稀釋、提取,通過HLB固相萃取柱凈化,液相色譜-質譜聯用儀分析的檢測方法。此方法前處理簡單、快速,操作簡便易行,且準確,靈敏度高,重現性好,檢出限低,能夠滿足大批量蜂蜜樣品中9種苯并咪唑類、新煙堿類農藥同時檢測的要求。
