AKT/FOXO1信號通路在心力衰竭小鼠骨骼肌萎縮中的作用
2018-11-01 08:12:28張聰聰陳博雅程乃萱
心肺血管病雜志 2018年1期
關鍵詞:小鼠
張聰聰 陳博雅 程乃萱 杜 杰
心力衰竭(heart failure, HF)是多種心血管疾病(高血壓、心肌梗死、先天性心臟病、瓣膜病等)導致的心臟病理性重構的結果。HF患者會感到疲勞、乏力、嗜睡和身體活動能力下降, 及包括呼吸肌在內的多種骨骼肌功能異常的表現。身體80%的蛋白質由骨骼肌儲存,骨骼肌萎縮會導致全身性的營養不良,骨骼肌萎縮的程度能夠作為HF導致死亡的重要預測指標[1-2]。因此,HF時骨骼肌蛋白分解代謝的研究具有重要意義。本研究采用主動脈弓橫向結扎(transverse aortic constriction, TAC)8周,復制小鼠HF動物模型[3]。采用實時定量-聚合酶鏈式反應(realtime-PCR) 和Western blot 技術,檢測HF小鼠脛骨前肌內Atrogin-1 和MuRF1的mRNA及蛋白表達變化,并采用Western blot 技術,檢測其上游轉錄因子FOXO1以及激酶AKT的磷酸化水平和總蛋白水平進行測定,比較磷酸化蛋白表達與總蛋白的比率。通過觀察骨骼肌組織中上述指標的變化,從而探討HF時骨骼肌萎縮的可能分子機制,并為有效的多靶點干預提供理論依據。
材料與方法
1. 實驗動物 SPF級C57BL/6J小鼠20只,雄性,10周齡,體質量20~23g,由首都醫科大學附屬北京安貞醫院實驗動物房提供。購自北京華阜康生物科技股份有限公司,生產許可證號:SCXK(京)2014-0004,使用許可證號: SCXK(京)2015-0023。小鼠隨機分為兩組: 對照組(10只) 和HF模型組(10只)。
2. 主要試劑 反轉錄試劑盒(Promega,美國),realtime-PCR試劑盒(TaKaRa,日本),Atrogin-1,MuRF1一抗(Abcam,美國),FOXO1,p-FOXO1,AKT, p-AKT一抗(Cell Signaling Technology,美國),GAPDH一抗(中杉金橋,中國),DyLight 800標記山羊抗兔IgG二抗(LI-COR,美國)。
3. TAC誘導心力衰竭模型制備 HF組小鼠使用1%戊巴比妥麻醉(70mg/kg體質量),從頸部剪開皮膚,游離甲狀腺,剪開胸骨,游離胸腺及主動脈弓周圍結締組織,充分暴露主動脈弓。使用5~0棉線結扎主動脈弓,以27號針為墊針結扎。結扎后撤出27號針頭,縫合胸骨和皮膚。對照組小鼠僅麻醉后剪開頸部皮膚剪開胸骨并暴露主動脈弓后進行縫合。所有小鼠均普通飲食飼養,自由進食、進水。分別于術前,術后1周,2周,4周,8周進行小鼠稱重。術后1周,通過小動物超聲(VIVO,美國)檢測主動脈弓流速,以流速≥2 500mm/s為模型成功標準。術后8周,測量心功能之后收取小鼠心臟組織,用10%中性甲醛固定后用于病理分析。同時無創游離小鼠雙側脛骨前肌,并進行稱重,一側用10%中性甲醛固定后用于病理分析,另一側用液氮凍存用于mRNA和蛋白的提取。
4. 小鼠心功能測定 于TAC術后8周,通過小動物超聲進行小鼠心功能測定,主要檢測指標如下:室間隔厚度(interventricular septum,IVS)、左心室后壁厚度(left ventricular posterior wall thickness,LVPW)、左心室舒張末期內徑(left ventricular end-diastolic dimension, LVEDD)、左心室收縮末期內徑(left ventricular end-systolic dimension, LVEDS)、左心室舒張末期容積(left ventricular end-diastolic volume, LVESV)、左心室收縮末期容積(left ventricular end-systolic volume, LVEDV)、左心室射血分數(left ventricular ejection fraction,LVEF)、左心室縮短分數(left ventricular fractional shortening,LVFS)。
5. 心臟組織、骨骼肌組織病理切片的制備方法同前[4]。取出已固定的心臟組織和脛骨前肌,常規石蠟包埋,4μm切片。HE染色:切片經二甲苯和梯度乙醇脫蠟至水,蘇木素染色3min,自來水沖洗;鹽酸乙醇分化30s;自來水返藍5min;置伊紅染色液2min;常規脫水,透明,封片。WGA染色:切片經二甲苯和梯度乙醇脫蠟至水,檸檬酸緩沖液(pH=6.0)抗原修復90s,1 x PBS緩沖液洗3遍,羊血清封閉液室溫封閉30min,WGA工作液(10ug/mL, Sigma,美國)37℃,孵育60min,1 x PBS緩沖液洗3遍,DAPI封片。使用電子熒光顯微鏡(Nikon,日本)進行病理圖像采集,使用NIS Br 3.0軟件進行圖像分析。
6. realtime-PCR檢測小鼠脛骨前肌組織中Atrogin-1和MuRF1的mRNA水平 取相同質量50 mg 的脛骨前肌放入勻漿器內,加入1 mL Trizol(Invitrogen,美國)提取RNA,用Nanodrop 2 000測定RNA 的濃度和OD260 /280 比值。各樣本均按5μg RNA 相對應的體積(n μL) 根據反轉錄試劑盒說明書進行逆轉錄,轉錄體系為20 μL 體系,將混合好的液體放置在PCR儀中,反應條件為42℃, 60min;95℃, 5min。將反轉好的cDNA按照realtime-PCR 說明書進行半定量PCR,使用Bio-RAD CFX Connect進行反應,反應條件為95℃, 2min;95℃,15s;60℃, 30min(40個循環);60℃, 5s,每個循環增加0.5℃~95℃。以GAPDH作為內參。基因的表達水平以得到的相應的Ct 值用2 (Ct GAPDH-Ct 目標基因)法進行計算。引物序列分別為: Atrogin-1∶5’- CGGGCTTCCTTGAGTGTCTT-3’(上游),5’- GGCTCTCCTAAGGTCCCAGA-3’(下游);MuRF-1∶5’- AAACTTGTGGA GACCGCCAT-3’(上游),5’- TCTTGATGAGCTGCTTGGCA-3’(下游);GAPDH:5’- CATGGCCTTCCGTGTTCCTA-3’(上游),5’-GCGGCACGTCAGATCCA-3’(下游)。
7. Western blot 測定小鼠脛骨前肌組織中相關蛋白的表達 取50mg脛骨前肌用300μL T-PER組織裂解液(含1x 0.5M EDTA, 1x蛋白酶抑制劑,1x磷酸酶抑制劑)(Thermo,美國)進行裂解,12 000r/min,離心15min,后吸取上清。使用BCA蛋白濃度測定試劑盒測定樣品總蛋白濃度。將100μg蛋白與5x蛋白上樣緩沖液進行混合后,100℃變性5 min。進行聚丙烯酰胺凝膠電泳,然后蛋白電轉移至硝酸纖維素膜,用5%脫脂奶粉溶液封閉2h,再依次孵育稀釋后的一抗Atrogin-1(兔多抗,1∶1 000),MuRF1(兔多抗,1∶1 000),FOXO1(兔多抗,1∶1 000),p-FOXO1(兔多抗,1∶1 000),AKT(兔多抗,1∶1 000), p-AKT(兔多抗,1∶1 000),GAPDH (鼠單抗,1∶1 000),4℃孵育過夜。洗滌。敷二抗(1∶1 000稀釋),室溫孵育1小時,稍洗。使用Odyssey 儀器進行熒光掃描,并分析條帶灰度。
8.統計學方法 應用GraphPad Prism 5 統計軟件對實驗數據進行統計學分析。計量數據以均數±標準差表示,采用獨立樣本的t檢驗。以P<0.05為差異有統計學意義。
結 果
1.HF 模型的結果 (1)體質量變化:TAC 術前和術后2周,4周時實驗組和對照組小鼠的平均體質量差異無統計學意義,TAC 6周時實驗組小鼠體質量明顯低于對照組小鼠(P<0.05),TAC 8周時實驗組小鼠體質量明顯低于對照組(P<0.01),且體質量下降已超過預計值的10%,出現營養不良改變。結果見圖1。
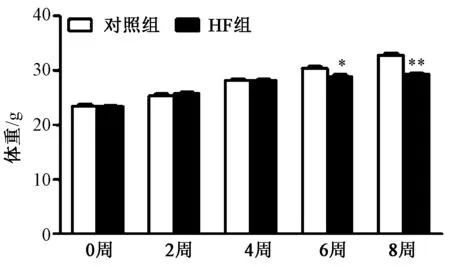
圖1 小鼠體質量變化 注:與對照組相比,*P<0.05,**P<0.01
(2)心功能變化:TAC術后8周時使用小動物超聲檢測兩組心功能各項指標,其中HF組的EF值和FS值較對照組顯著下降(P<0.01,表1)。
(3)心臟組織形態學改變:心臟組織HE染色可見HF組小鼠心臟室壁厚度較對照組增加,心室內徑增加。WGA染色觀察心肌細胞的面積發現HF組心肌細胞的面積大于對照組(P<0. 01,圖2)。
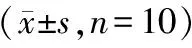
表1 兩組小鼠心功能比較
注:LVM:左心室質量

圖2 小鼠心肌病理檢測 A:HE染色(放大倍數40X,標尺500μm);A心動衰竭組:心臟左心室室壁厚度的測量數據,與對照組相比,*P<0.05;B:WGA染色(放大倍數400X,標尺50μm),B心動衰竭組:根據WGA染色心肌細胞的橫截面面積,與對照組相比,** P<0.01
(4)骨骼肌組織形態學觀察:HF組脛骨前肌重量和脛骨長度比顯著低于對照組。HE染色可見HF組肌纖維橫截面積較對照組減小,經過WGA染色統計分析肌纖維面積可見HF組肌纖維橫截面面積顯著小于對照組,每高倍鏡視野下肌纖維數量計數顯著高于對照組。提示HF組骨骼肌發生萎縮。見圖3~4。
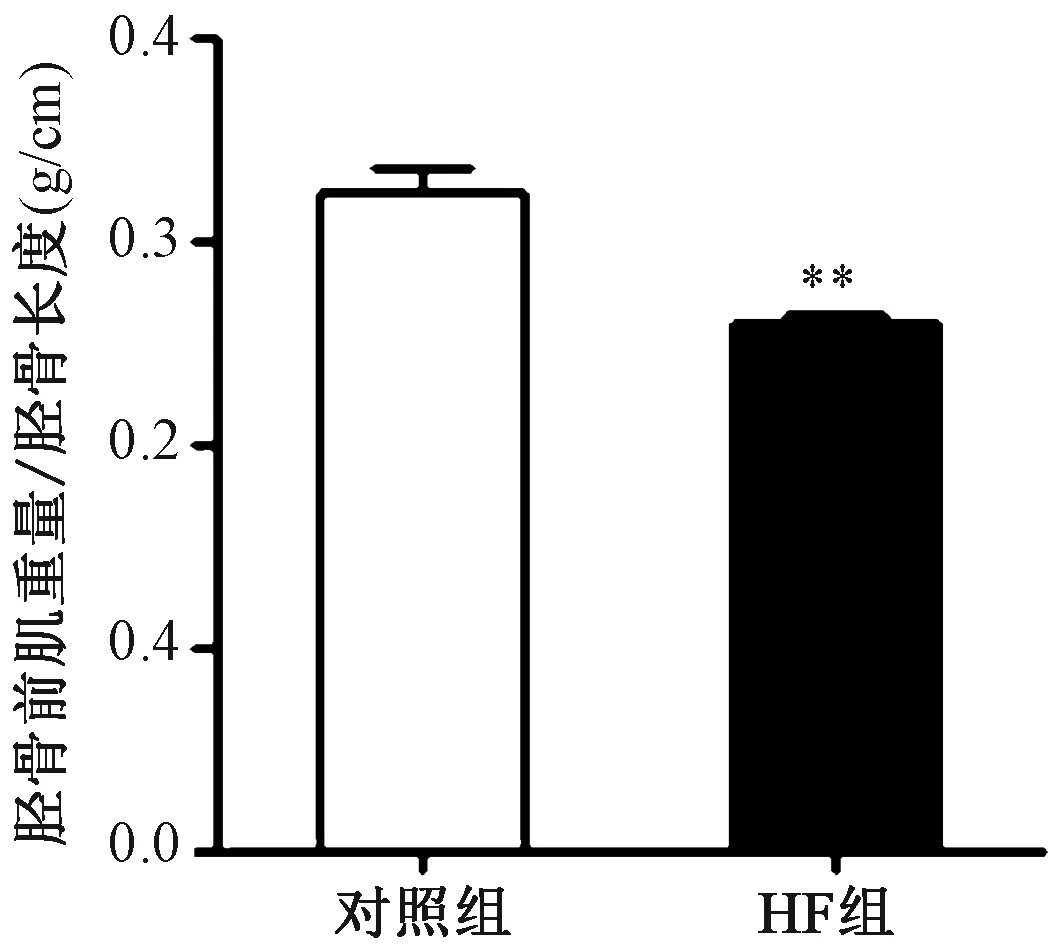
圖3 小鼠脛骨前肌和脛骨長度比值, 與對照組相比,**P<0.01
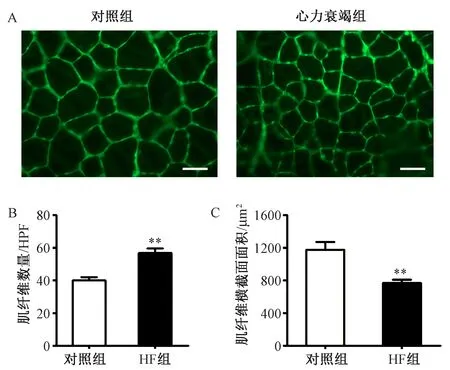
圖4 小鼠脛骨前肌肌纖維病理分析 A:WGA染色(放大倍數X400,標尺50μm);B:每高倍視野(HPF)中肌纖維數量的統計;C:肌纖維面積的統計結果注:與對照組相比,**P<0.01
2.脛骨前肌中Atrogin-1和MuRF1 的mRNA 和蛋白表達變化 脛骨前肌realtime-PCR的結果顯示,HF組脛骨前肌中Atrogin-1的mRNA水平較對照組增加約3.8倍 (P<0.01); HF組脛骨前肌中MuRF1的mRNA水平較對照組增加7.2倍 (P<0.01,圖5)。Western Blot的結果顯示,Atrogin-1 的蛋白相對表達量HF組較對照組明顯增加,兩組比較差異有統計學意義(P<0.01); MuRF1 的蛋白表達HF組較對照組增加(P<0.05,圖6)。

圖5 兩組小鼠脛骨前肌中Atrogin-1和MuRF-1的mRNA表達水平 注:與對照組相比,**P<0.01
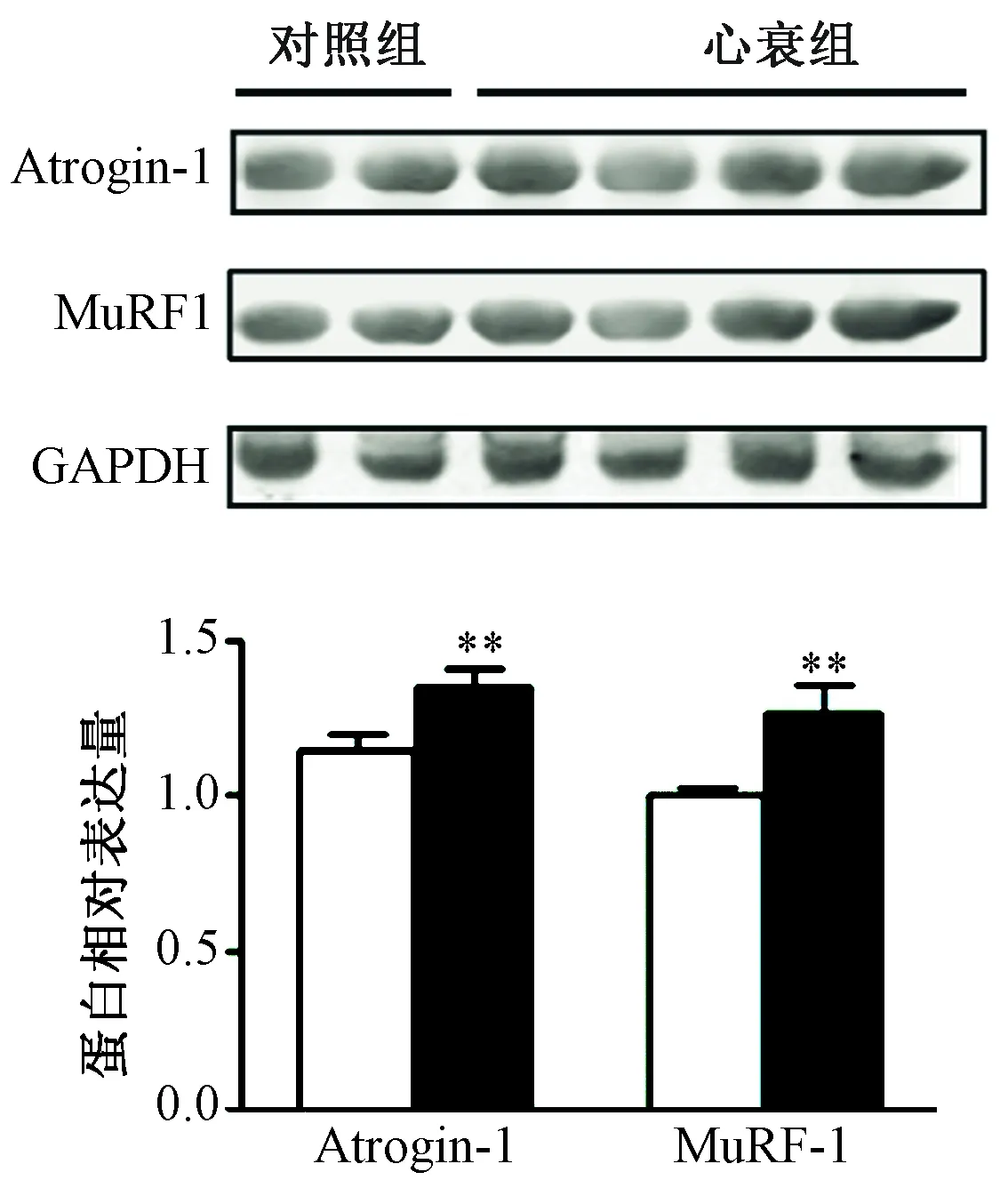
圖6 兩組小鼠脛骨前肌中Atrogin-1和MuRF-1的蛋白表達水平 注:與對照組相比,**P<0.01
3.脛骨前肌中AKT/FOXO1信號通路的活化變化 Western blot 分析結果顯示,AKT和FOXO1總蛋白的表達量在HF組和對照組差異無統計學意義。磷酸化AKT和磷酸化FOXO1的水平在HF組顯著增加,兩組比較差異有統計學意義(P<0.05)。p-AKT/AKT和p-FOXO1/ FOXO1的比值在HF組顯著增加,兩組比較差異有統計學意義(P<0.05,圖7)。
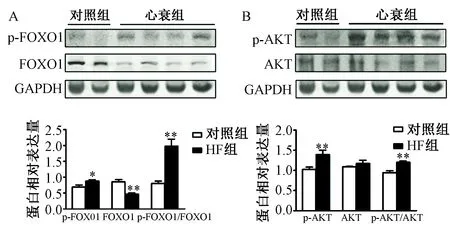
圖7 A:兩組小鼠脛骨前肌中p-FOXO1和FOXO1的蛋白表達水平;B:兩組小鼠脛骨前肌中p-AKT和AKT的蛋白表達水平,注:與對照組相比,*P<0.05; 與對照組相比,**P<0.01
討 論
根據中國心力衰竭注冊登記記錄統計,中國35~74歲人群慢性心力衰竭患病率為0.9%,并呈上升趨勢,其中住院心力衰竭患者的病死率約為5.3%[5]。心力衰竭患者骨骼肌萎縮的程度直接影響疾病的預后情況[2]。心力衰竭引起骨骼肌病變的主要表現為:①重量減輕;②結構改變, 如肌纖維面積變小、I型纖維向Ⅱ型纖維轉變、線粒體減少;③能量代謝的改變,蛋白水解、糖酵解增加;④細胞因子及氧化標記物表達增加等[6]。心力衰竭患者發生骨骼肌萎縮是一種多因素、多種分子生物學機制共同參與的復雜過程[7],其最終歸結為骨骼肌蛋白合成和降解之間的不平衡,蛋白降解途徑增加程度大于蛋白合成,導致骨骼肌萎縮。我們在本研究中使用TAC 8周建立小鼠HF模型,發現小鼠的骨骼肌重量減輕,肌纖維面積減小,提示小鼠HF導致骨骼肌萎縮的模型成功,可以為研究小鼠骨骼肌萎縮的分子機制提供基礎。
國內外的研究顯示,骨骼肌萎縮過程中的蛋白質的降解途徑主要包括溶酶體途徑、泛素-蛋白酶體途徑和Ca2+依賴性途徑等[8],其中泛素-蛋白酶體途徑最為重要。在肌肉中特異性表達的E3 泛素連接酶主要為MAFbx (muscle atrophy F-box,又稱為Atrogin-1)和MuRF-1 (muscle RING finger 1),這兩個分子屬于成肌分化抗原(myogenic differentiation antigen, MyoD)降解過程中的核蛋白[9]。在發生HF的心臟組織中Atrogin-1和MuRF-1可出現高表達[10-11]。Li 等的實驗證明,腫瘤壞死因子 (tumor necrosis factor,TNF)-α可通過激活絲裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK) p38亞基來調節Atrogin-1的表達[12]。細胞因子胰島素樣生長因子(insulin-like growth factor,IGF)-1的表達可以抑制Atrogin-1和MuRF-1的表達從而抑制骨骼肌萎縮的發生[13]。先前有研究顯示在慢性阻塞性肺疾病導致的骨骼肌萎縮過程中骨骼肌中的Atrogin-1和MuRF-1的表達就顯著增加[14]。我們在本研究中發現HF小鼠的骨骼肌中兩種泛素連接酶Atrogin-1和MuRF-1的表達顯著增加,這兩個分子可能引起骨骼肌中蛋白質的泛素化增加, 促進其被泛素蛋白酶體系統降解,發生骨骼肌萎縮。
轉錄因子FOXOs 家族是一類擁有翼狀螺旋結構的蛋白,主要包括FOXO-1 和FOXO-3 等。FOXOs 蛋白通過其C末端的轉錄激活結構域與靶基因的轉錄啟動子區域結合促進靶基因的表達[15]。當FOXOs被上游的信號分子如AKT等磷酸化后使其由細胞核轉運至細胞質,進而被泛素蛋白酶體系統降解,表達降低,導致其轉錄活性下調,從而抑制其對靶基因的轉錄調控作用。研究顯示FOXOs可以通過調控自噬相關基因的轉錄引起骨骼肌細胞的自噬,同時可以促進肌細胞中MyoD的表達影響肌纖維的分化[16-17]。最近的研究也顯示FOXO1可以直接結合至與骨骼肌萎縮密切相關的Atrogin-1 和MuRF1的轉錄啟動子區域,從而調節Atrogin-1 和MuRF1的表達[18]。研究顯示骨骼肌特異性敲除FOXO1基因將抑制廢用性骨骼肌萎縮或去神經性骨骼肌萎縮的發生[19-20]。我們在本研究中發現HF組骨骼肌組織中FOXO1的磷酸化水平增加,同時FOXO1總蛋白水平下降,提示在心力衰竭導致骨骼肌萎縮的過程中存在負向調控分子導致FOXO1的轉錄調控能力下降從而抑制骨骼肌萎縮的發生。
AKT是一種絲/蘇氨酸蛋白激酶,在磷脂酰肌醇依賴的蛋白激酶(phosphoinositide 3-kinase, PI3K)的協同作用下,磷脂酰肌醇2磷酸(PIP2)和磷脂酰肌醇3磷酸(PIP3)與胞漿內AKT 結合,AKT轉位 到質膜,并促進Ser473 和Thr308 位點磷酸化。Ser473或/和Thr308 位點的磷酸化是AKT 激活的必要條件,活化狀態下的AKT 使轉錄因子FOXO1 磷酸化后從細胞核中移出,并在細胞漿中被泛素蛋白酶體所降解,從而抑制其轉錄調節活性[21]。Atrogin-1 和MuRF1 受FOXOs 轉錄因子家族的轉錄調節,而AKT 可使FOXOs基因轉錄調節功能失活。本研究中發現HF小鼠的骨骼肌中AKT和FOXO1的磷酸化顯著高于對照組,提示骨骼肌中存在激活AKT下調FOXO1的轉錄活性,從而抑制Atrogin-1 和MuRF1表達抑制骨骼肌萎縮的負性調控因子。另一方面,磷酸化的AKT可激活哺乳動物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)和真核翻譯起始因子4E結合蛋白1(e IF4E-binding protein 1,4E-BP1)、核糖體S6激酶1(ribosome protein subunit 6 kinase 1, S6K1)信號通路,并阻礙抑制蛋白質合成因子糖原合成酶激酶3β (glycogen synthase kinase-3β, GSK-3β)活性,促進蛋白合成[22-23],啟動負向調控機制。
綜上所述,心力衰竭的骨骼肌中蛋白降解相關基因Atrogin-1 和MuRF1的表達顯著上調促進骨骼肌萎縮的形成。同時骨骼肌中AKT/FOXO1信號通路活化,啟動負向調控機制,可能發揮抑制骨骼肌萎縮的作用,成為機體的一個代償機制。但是具體何種分子激活AKT/FOXO1信號通路尚需進一步的探索。本研究的發現為治療心力衰竭骨骼肌萎縮提供一個新思路,即干預AKT/FOXO1 信號通路上的某些靶點使其進一步激活,從而達到抑制骨骼肌萎縮的目的。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
