基于通路分析的重型痤瘡全基因組關聯研究
2018-10-12 02:37:14楊健康馮家祺張平曹光瓊何黎吳文娟
中華皮膚科雜志 2018年9期
楊健康 馮家祺 張平 曹光瓊 何黎 吳文娟
671000云南,大理大學基礎醫學院(楊健康);昆明醫科大學第一附屬醫院皮膚科(馮家祺、何黎、吳文娟),體檢中心(曹光瓊);云南曲靖市第一人民醫院檢驗科(張平)
痤瘡是一種常見的皮膚疾病,重型痤瘡是痤瘡中最嚴重的一類,它主要表現為廣泛分布的炎癥性皮損,經常導致患者的心理問題[1?2]。遺傳因素在重型痤瘡的發生中發揮重要作用[3]。全基因組關聯研究(GWAS)是一種發現疾病易感基因的有效方法[4?5]。針對中國人群重型痤瘡的GWAS研究曾發現2個新的易感基因[6]。但是,基因通常通過基因通路的形式在疾病的發生、發展中發揮作用[7]。因此,有必要進行基因通路分析,研究基因通路與疾病的關系,從而為全基因組關聯研究提供有益補充。為了研究基因通路與重型痤瘡的關系,我們進行重型痤瘡全基因組通路分析,希望提高對重型痤瘡發病機制的認識。
材料與方法
一、 數據
分析來源于重型痤瘡全基因組關聯研究的單核苷酸多態性(SNP)芯片數據[6]。采用美國Illumina公司HumanOmniZhongHua?8型芯片,于安徽醫科大學皮膚病學教育部重點實驗室行芯片掃描。數據集包括1 056例重型痤瘡患者和1 056例健康對照的芯片數據,每個樣本檢測900 015個SNP位點。所有的重型痤瘡患者都經皮膚科副主任或主任醫師診斷為4型(Pillsbury分級系統):除顏面及胸背部泛發的粉刺、丘疹、膿皰外,皮損炎癥重,并有囊腫、結節和瘢痕。本研究經昆明醫科大學第一附屬醫院醫學倫理委員會批準,患者及健康對照均簽署知情同意書。標本由昆明醫科大學第一附屬醫院、云南曲靖市第一人民醫院、安徽醫科大學第一附屬醫院等全國三十余家醫院合作收集完成。
二、 方法
1.數據質控:對原始的SNP芯片數據進行嚴格的質量控制,以去除不合格的樣本和SNP位點。首先對標本進行質控,排除檢出率(call rates)<98%的樣本,剩下合格的1 031份病例樣本和1 031份健康對照樣本。接著對SNP位點進行質控,①排除所有樣本中檢出率<98%的SNP位點;②排除所有樣本中最小等位基因頻率(MAF)<0.01的SNP位點;③排除健康對照樣本中嚴重偏離哈迪-溫伯格平衡(P< 1× 10?4)的SNP位點。經過質控,共篩出合格的SNP位點809 305個。
2.SNP位點分析:使用Plink v1.90軟件[8]對SNP位點進行logistic回歸分析。使用Q?Q圖評估群體分層的大小及對結果的影響。使用Haploview v4.2軟件做全基因組曼哈頓圖。
3.基因分析:使用VEGAS軟件計算[9],采用蒙特卡羅(Monte Carlo)法統計基因與重型痤瘡的相關性,P值代表基因與重型痤瘡相關的顯著性水平。為了包括基因調控區的SNP位點,將基因的范圍定義為編碼序列(CDS)和20 kb的5′UTR及20 kb的3′UTR。接著將SNP定位到基因上。計算P值時,考慮了同一基因上所有SNP位點的影響,并且使用HapMap CHB和JPT的數據進行連鎖不平衡校正。
4.通路分析:使用在線工具WebGestalt進行基因通路分析[10]。該基因通路為KEGG數據庫定義的基因通路。為了減少通路大小對結果的影響,僅對包括10~200個基因的通路進行分析。采用超幾何分布檢驗計算每條通路的P值,使用錯誤發現率(FDR)對得到的結果進行多重檢驗校正,校正后P<0.05視為通路與重型痤瘡相關。
結 果
一、 SNP位點分析結果
40 920個SNP位點的P值小于0.05,見全基因組曼哈頓圖(圖1)。從圖2可以看出所有SNP位點對應的P值整體上沒有偏離零假設分布,提示病例對照樣本匹配良好,人群分層因素對結果影響較小,與重型痤瘡顯著相關的SNP位點應該是真實的相關信號。
二、 基因分析結果
所有基因中,919個對應的P值小于0.05,其中12個P值小于0.001,見表1。
三、 通路分析結果
發現5條顯著的基因通路(P<0.05),見表2。根據KEGG通路分類,可以將這5條通路分為代謝(1條)、內分泌系統(1條)、癌癥(1條)、免疫系統(2條)。
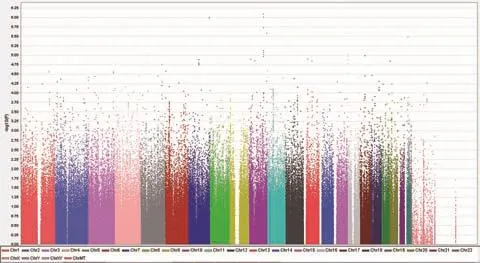
圖1 全基因組關聯分析曼哈頓圖 不同的顏色分別代表23對染色體;縱坐標為每個單核苷酸多態性(SNP)位點的-log10(P)值,該值越大代表該位點的等位基因頻率在病例組與對照組間的差異越顯著
討 論
查詢PubMed數據庫和CNKI數據庫,截至2018年6月,未檢索到重型痤瘡全基因組通路研究的相關文獻,本研究可能是國際上第一個重型痤瘡的全基因組通路研究,將有利于加深我們對于重型痤瘡發病機制的認識。本研究發現的通路中有2個與免疫反應和炎癥相關,分別是丙型肝炎和高親和力IgE受體信號通路。這和目前普遍認為重型痤瘡是炎癥性疾病的認識一致[11?12]。研究認為,炎癥反應會刺激毛囊皮脂腺導管角化過度,從而影響皮脂的正常排出[13]。
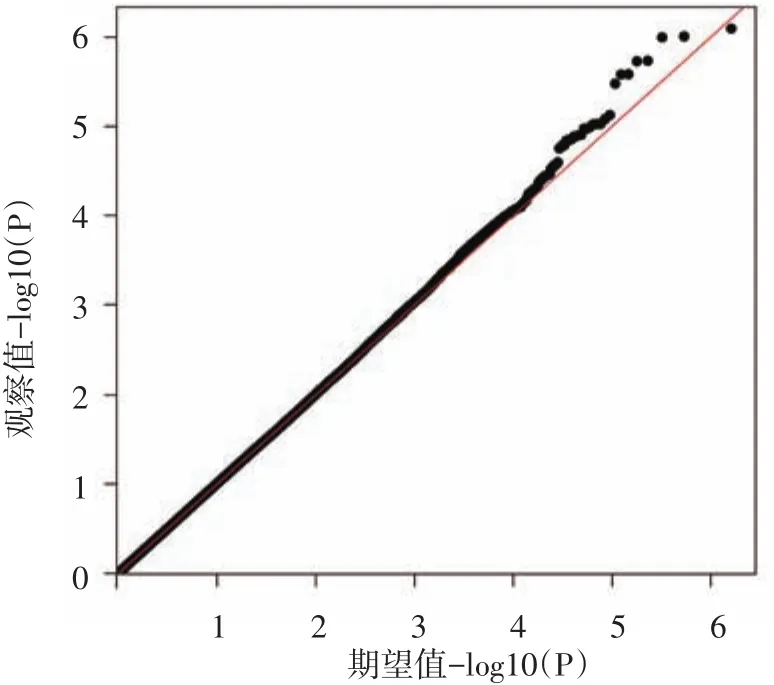
圖2 全基因組關聯分析Q-Q圖 橫坐標為809 305個單核苷酸多態性(SNP)位點-log10(P)值的期望值,縱坐標為-log10(P)值的觀察值,圖中觀察值與期望值沒有偏離零假設分布,提示病例對照樣本匹配良好,人群分層因素對結果影響較小
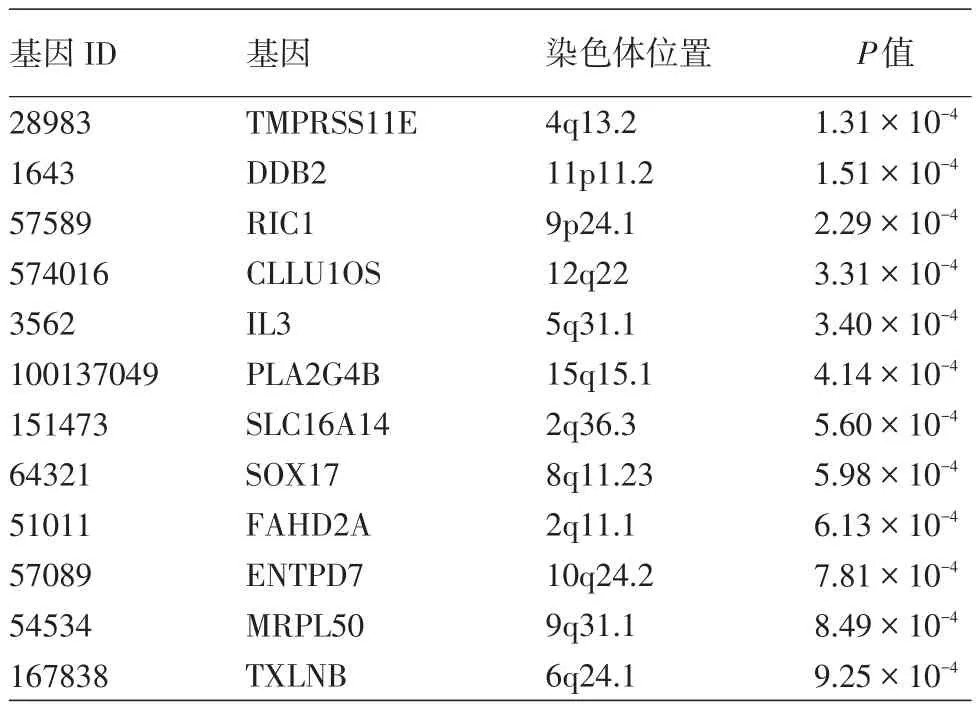
表1 基因分析發現的12個與重型痤瘡關系最密切的基因
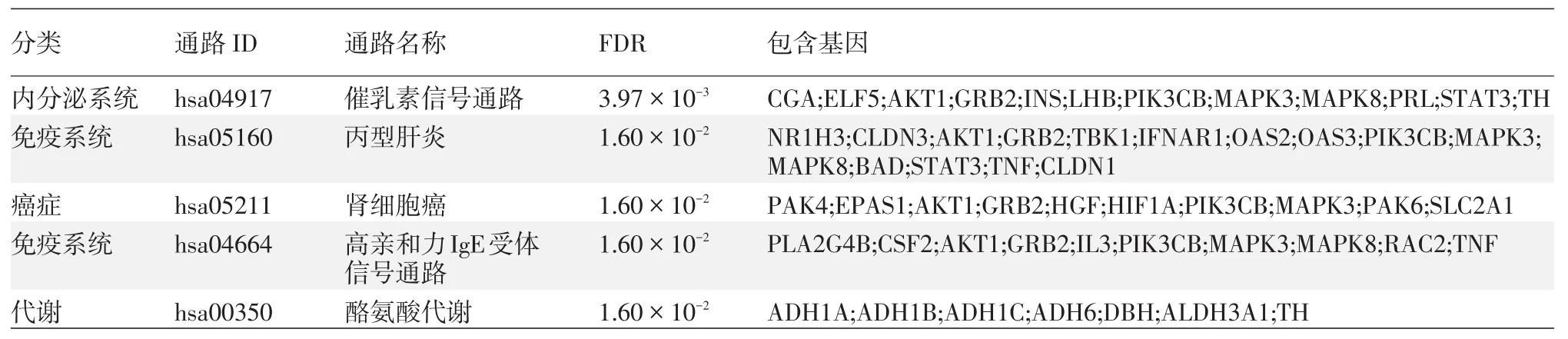
表2 通路分析方法發現的與重型痤瘡相關的5條基因通路
本研究發現一些新的基因通路可能與重型痤瘡相關,提示除了炎癥、免疫相關通路以外,其他通路可能也參與重型痤瘡的發生和發展。比如酪氨酸代謝通路,酪氨酸對于神經遞質多巴胺的合成也發揮重要作用[14]。多巴胺是可以使人產生快樂的神經遞質[15]。一項針對新加坡青少年進行的研究發現,痤瘡的嚴重程度與精神壓力呈正相關[16]。因此,酪氨酸代謝可能對于重型痤瘡患者的精神壓力也有重要的影響[17]。催乳素信號通路和腎細胞癌通路與痤瘡的關系未見相關文獻報道,需要進一步的功能研究來明確。
本次基因分析發現12個基因的P值小于0.001,其中包括重型痤瘡全基因組關聯研究發現的DDB2基因。DDB2基因編碼的蛋白是一個新發現的與雄激素受體有相互作用的蛋白,可能通過泛素化使雄激素受體蛋白被蛋白酶體降解[18]。其他基因如CLLU1OS、IL3與炎癥相關,IL3在激活與調節免疫細胞和介導T、B細胞增殖與分化及痤瘡的炎癥反應中起重要作用[19]。
本研究結果只是針對中國人群重型痤瘡GWAS研究的全基因組通路分析,部分結果也可能存在假陽性的問題,有待新的重型痤瘡GWAS研究對這些結果進行驗證。我們發現一些可能與重型痤瘡發病相關的基因通路,有望為后續的研究提供一些思路。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
當代陜西(2021年17期)2021-11-06 03:21:36
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年18期)2018-11-14 01:48:24
學苑創造·A版(2018年11期)2018-02-01 06:29:20
讀者(2017年5期)2017-02-15 18:04:18
山東工業技術(2016年15期)2016-12-01 05:31:22
