原發腎上腺NK/T細胞淋巴瘤皮膚侵犯一例
2018-08-03 13:27:28陳玲境韓睿程浩
中華皮膚科雜志 2018年7期
陳玲境 韓睿 程浩
310020杭州,浙江大學醫學院附屬邵逸夫醫院皮膚科
患者女,64歲,診斷腎上腺NK/T細胞淋巴瘤1年,四肢暗紅色浸潤性斑塊2個月。患者1年前因劇烈腹痛就診于杭州市中醫院,腹部CT發現雙側腎上腺腫塊,行左腎上腺切除術,結合免疫組化、病理,診斷為非霍奇金淋巴瘤,NK/T細胞淋巴瘤。10個月前于浙江大學醫學院附屬邵逸夫醫院就診,腹部CT示右側腎上腺巨大團塊。血常規、凝血功能、胸部CT檢查未見明顯異常。骨髓穿刺及組織病理檢查示骨髓組織增生活躍,T細胞受體γ、IgH及IgK基因重排陰性。給予6個周期化療:AspaMetDex方案(培門冬酶+甲氨蝶呤+地塞米松)1個周期4 d,GELOX方案(吉西他濱+門冬西酰胺酶+奧沙利鉑)1個周期5 d,CHOP+L方案(門冬酰胺酶+脂質體阿霉素+環磷酰胺+長春地辛+潑尼松)4個周期4 d,繼以1個周期放療。2個月前四肢皮膚出現暗紅色浸潤性斑塊,無疼痛瘙癢等,遂來我院皮膚科就診。體檢:四肢皮膚干燥脫屑,散在圓形、類圓形暗紅色斑塊,伴浸潤感,界限清楚(圖1)。實驗室檢查:白細胞2.0×109/L,紅細胞2.9×1012/L,血小板131×109/L,血紅蛋白94 g/L,淋巴細胞比例0.18。腹部CT:右腎上腺可見57 mm×18 mm低密度影。右小腿后側皮損組織病理檢查:角化過度,表皮菲薄,棘層海綿水腫,散在角化不良細胞,表皮內單個核細胞浸潤,基底層液化變性,并伴單個核細胞浸潤;真皮淺層水腫,血管周圍大量單個核細胞浸潤,可見噬色素細胞,膠原纖維水腫斷裂(圖2)。EB病毒編碼RNA(EBER)原位雜交陽性。免疫組化:CD56、胞質CD3、CD45RO、CD4、CD5、CD7、CD8、CD163、T細胞內抗原1、Ki67均陽性;CD20、CD30、粒酶B均陰性。
診斷:原發腎上腺NK/T細胞淋巴瘤皮膚侵犯。
治療:給予干擾素α-2b 300萬IU皮下注射隔日1次,地塞米松磷酸鈉20 mg靜脈滴注每日1次連續4 d,培門冬酶3 750 IU單次肌內注射,治療3個周期(每周期4 d),皮疹顏色減退,但腹部CT示兩腎新發多處低密度灶,診斷為雙腎淋巴瘤進展。
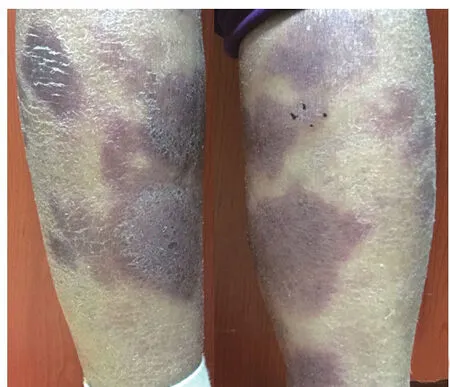
圖1 患者雙小腿散在暗紅色斑塊,伴干燥脫屑
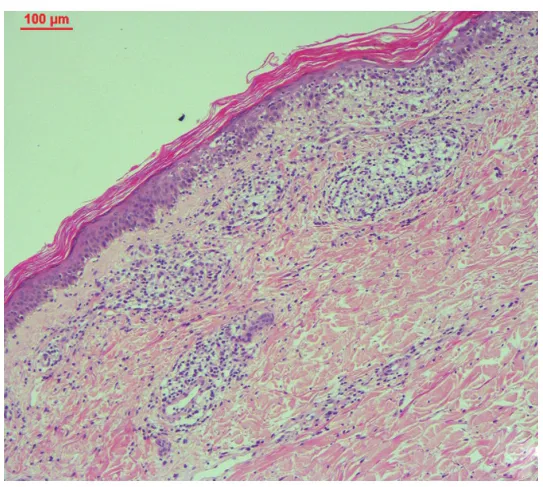
圖2 皮損組織病理
討論結外NK/T細胞淋巴瘤屬于非霍奇金淋巴瘤的一種特殊類型,具有以血管為中心的多形性淋巴細胞浸潤的特征,伴血管破壞和明顯壞死,以鼻腔及面部中線鼻竇、鼻咽、上腭、咽、喉多見,少數病例可原發于皮膚、腸道、睪丸、肺等部位。研究表明,NK/T細胞淋巴瘤與EB病毒感染密切相關[1-2]。本例患者四肢暗紅色浸潤性斑塊,組織病理可見血管周圍大量單個核細胞浸潤,表皮層和基底層可見單個核細胞,EBER原位雜交陽性,CD56、胞質CD3、CD45RO、T細胞內抗原1陽性,符合皮膚NK/T細胞淋巴瘤診斷。患者皮膚受累發生于腎上腺NK/T細胞淋巴瘤疾病后期,皮疹發于四肢遠端,鼻部和面部皮膚未受累,皮損組織與左腎上腺腫塊的病理及免疫組化結果一致,因此,診斷為腎上腺NK/T細胞淋巴瘤皮膚侵犯。本例需與蕈樣肉芽腫、皮下脂膜炎樣T細胞淋巴瘤、原發性皮膚侵襲性親表皮性CD8+細胞毒性T細胞淋巴瘤和皮膚原發NK/T細胞淋巴瘤鑒別。蕈樣肉芽腫斑塊期組織病理主要表現為真皮淺層大量慢性炎癥細胞呈帶狀浸潤,其中有多數腦回狀或核大而形態不規則的蕈樣肉芽腫細胞,瘤細胞呈親表皮性浸潤,表皮內可見Pautrier微膿腫,其免疫表型主要是CD4陽性,但CD8通常陰性。皮下脂膜炎樣T細胞淋巴瘤典型表現是一個或多個通常無痛的皮下結節或界限不清的硬化斑塊,可累及腿部、手臂、軀干和/或面部,組織病理顯示不典型淋巴細胞浸潤皮下脂肪小葉,但脂肪小葉間隔及上覆的真皮和表皮通常不受累,腫瘤細胞可表達CD3、CD8、粒酶B、T細胞內抗原1、穿孔素和T細胞受體α/β,但CD4和CD56陰性。原發性皮膚侵襲性親表皮性CD8+細胞毒性T細胞淋巴瘤主要表現為局限或播散的結節、腫瘤伴中心潰瘍及壞死,淋巴細胞有明顯親表皮性,可伴表皮角質形成細胞壞死、水皰甚至潰瘍,淋巴細胞異形性明顯,皮膚附屬器被侵犯、破壞,CD8+T淋巴細胞浸潤易侵犯并破壞血管,浸潤細胞表達CD3、CD8、βF1、CD45RA、粒酶 B、T細胞內抗原1、穿孔素,不表達CD45RO、CD2、CD5,一般不伴EB病毒感染。
由于該病較少見,尚無標準治療方案,臨床中具體治療方案的選擇缺少有力的證據支持。本例患者接受化療后,四肢皮疹消退,但雙腎淋巴瘤仍有進展。皮膚侵犯預示著較低的完全緩解率,疾病進展風險和死亡風險分別增加7.3倍和2.8倍,5年總生存率為21%,中位生存時間<12個月[3]。
