天然氣水合物注二氧化碳置換程度 計算新模型
2018-07-10 11:40:10關(guān)富佳
天然氣與石油 2018年3期
關(guān)鍵詞:效率
張 杰 陳 花 關(guān)富佳 肖 娜
長江大學(xué)石油工程學(xué)院, 湖北 武漢 430100
0 前言
目前,關(guān)于天然氣水合物開采方法的研究均處于理論論證和實驗?zāi)M階段,研究方法主要包括熱力學(xué)方法[1]、降壓開采方法[2]、注抑制劑方法[3]、地面分解方法[4]、置換開采方法[5]等。熱力學(xué)方法普遍存在導(dǎo)熱系數(shù)較低的情況,開采過程中能量損失較大[6];降壓開采方法在降壓開采過程中,會導(dǎo)致水合物礦藏孔隙被壓縮,滲流能力下降,加之水合物自保護效應(yīng)的作用,導(dǎo)致水合物礦藏開發(fā)程度較低[7];注抑制劑方法中傳統(tǒng)的熱力學(xué)抑制劑容易造成地層污染,而新型的動力學(xué)抑制劑主要防止天然氣管道輸送過程中水合物的二次生成[8];地面分解方法實施過程中,對海底淺層地質(zhì)的挖掘很大程度上會對海底淺層疏松地層造成嚴(yán)重的地質(zhì)災(zāi)害[9]。
通過注二氧化碳置換開采天然氣水合物,開采過程中沒有水合物籠形結(jié)構(gòu)的破壞與重組,實驗研究發(fā)現(xiàn)置換過程沒有水合物的分解,置換過程在原位發(fā)生,沒有硬度丟失,不會造成地層失穩(wěn)情況的發(fā)生,顯示應(yīng)用前景較好。考慮到當(dāng)前技術(shù)條件下,二氧化碳置換技術(shù)存在置換效率較低的問題,提出置換效率受溫度影響的新模型,通過合理調(diào)控置換體系溫度,提高置換反應(yīng)程度。
1 考慮溫度的二氧化碳置換模型
李遵照等人[10]在等壓實驗條件下對比不同置換溫度的甲烷水合物的分解量隨時間變化關(guān)系,指出升溫能促進置換反應(yīng)的進行。王金寶等人[11]在等壓條件下利用氣相色譜儀測定兩種不同置換溫度下甲烷氣體的產(chǎn)出量,結(jié)果顯示升溫增大氣相組分中甲烷占比,表明置換程度隨溫度升高而升高。楊光等人[12]在對置換的熱力學(xué)進行研究時,指出二氧化碳水合物的熱穩(wěn)定性較甲烷水合物的熱穩(wěn)定性更好,在一定溫度范圍內(nèi),升溫能提高置換反應(yīng)進行程度。祁影霞等人[13]定義置換效率為采樣氣體中甲烷的摩爾分?jǐn)?shù),指出隨著溫度上升,置換效率增大。王小文等人[14]指出置換過程二氧化碳?xì)怏w會與沉積物中自由水反應(yīng)生成水合物,導(dǎo)致置換效率高于實際值。孫建業(yè)[15]在置換實驗中指出升溫對快速置換階段置換效率影響較小,對緩慢反應(yīng)階段置換效率影響較大。
本文將置換效率定義為置換出的甲烷量與參與合成反應(yīng)的甲烷量的比值,避免因二氧化碳?xì)怏w與原位自由水反應(yīng)生成二氧化碳水合物而造成置換效率不準(zhǔn)確的情況,重點分析新模型溫度對水合物合成和置換的影響,在保證甲烷氣體置換量的情況下更多地儲存二氧化碳?xì)怏w。
實驗條件下,甲烷水合物的合成發(fā)生在填砂管中,現(xiàn)對水合物合成情況進行理論推導(dǎo),取甲烷相為少數(shù)相,默認(rèn)合成為SI型水合物且水合物不可壓縮,依據(jù)等容合成原理有如下等式:
(1)
式中:VW1為反應(yīng)初始注水體積,通過平流泵測定,cm3;Z1為初始反應(yīng)溫壓條件下甲烷氣體壓縮因子;T1為合成反應(yīng)初始體系溫度,K;VG1為反應(yīng)初始甲烷標(biāo)況下氣體體積,cm3;p1為水合物合成反應(yīng)開始前壓力,MPa;Z2為反應(yīng)結(jié)束后甲烷氣體壓縮因子;T2為合成反應(yīng)結(jié)束后體系溫度,K;VG2為反應(yīng)結(jié)束后剩余的甲烷氣體標(biāo)況下體積,cm3;p2為水合物合成反應(yīng)結(jié)束后壓力,MPa;Vh為反應(yīng)生成的水合物體積,cm3;VW2為反應(yīng)結(jié)束后剩余水相體積,cm3。
默認(rèn)甲烷氣體完全占據(jù)小籠和中籠結(jié)構(gòu),即合成SI型甲烷水合物按照1∶164進行儲氣,按照1∶0.8進行儲水[16]。同時,默認(rèn)甲烷在水溶液中的溶解不會改變水溶液體積。同時反應(yīng)體系前后質(zhì)量不變,默認(rèn)水不可壓縮,對式(1)進行變換得:
(2)
式中:Vm為溶解在水中甲烷氣體標(biāo)況下體積,cm3。
關(guān)于氣體在液體里的溶解度通過Henry’s Law進行計算:
X(g)?X(aq)
(3)
(4)
式中:X(g)為氣體平衡濃度,mol/L;X(aq)為氣體在水中的平衡濃度,mol/L;pH為一定壓力下氣體的平衡分壓,Pa;KH為一定溫度下亨利常數(shù),查表得出,Pa·L/mol。
相對于水合物合成反應(yīng)消耗氣量而言,甲烷氣體在水中的溶解可以忽略,初始溫壓條件下甲烷注入體積可以通過孔隙體積和注水量的差值計算得出,對式(2)進行變形,得到反應(yīng)結(jié)束后剩余的甲烷氣體的體積:
(5)
將式(5)代入到式(2)進行變形,獲得如下等式:
(6)
式中:ΔV為參與反應(yīng)標(biāo)況下甲烷氣體量,cm3。
單因素分析式(6)顯示當(dāng)水合物合成反應(yīng)體系溫度T1越低,獲得的參與反應(yīng)甲烷氣量越多,即合成更多的水合物。
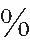
(7)
式中:Z3為置換反應(yīng)開始前溫壓條件下二氧化碳?xì)怏w壓縮因子;VG3為注入二氧化碳標(biāo)況下體積,cm3;p3為注入二氧化碳?xì)怏w結(jié)束后體系壓力,MPa。
求得此時體系壓力:
(8)
分析式(8),可以通過改變二氧化碳注入體積實現(xiàn)改變置換反應(yīng)壓力條件。此時,設(shè)定置換反應(yīng)初始溫度T3,并通過恒溫設(shè)備維持置換反應(yīng)體系溫度不變,當(dāng)反應(yīng)體系壓力穩(wěn)定維持10 min不再變化,默認(rèn)反應(yīng)完成。此時壓力維持在p4不變,考慮到甲烷和二氧化碳分子均與水形成SI型水合物,默認(rèn)置換反應(yīng)過程中水合物相不發(fā)生體積變化,只有壓力變化引起壓縮因子的變化,根據(jù)反應(yīng)體系容積不變原理,獲得如下等式:
(9)
式中:VG4為置換結(jié)束后標(biāo)況下甲烷體積,cm3;p4為置換反應(yīng)結(jié)束后體系壓力,MPa;VG5為置換結(jié)束后標(biāo)況下二氧化碳體積,cm3;Z4為置換反應(yīng)結(jié)束后p4壓力條件下甲烷壓縮因子;Z5為置換反應(yīng)結(jié)束后p4壓力條件下二氧化碳壓縮因子。
考慮到置換過程中微觀機理,又有如下等式:
VG3=0.75ΔV=VG4+VG5+VC
(10)
式中:VC為置換過程溶解在水中標(biāo)況下二氧化碳?xì)怏w體積,不可忽略,cm3。
二氧化碳?xì)怏w在水中溶解量可以依據(jù)化學(xué)勢相等計算得出:
(11)
二氧化碳溶液中溶質(zhì)化學(xué)勢為:
(12)
當(dāng)溶解平衡,氣液化學(xué)勢相等,求得氣體溶解量。默認(rèn)置換反應(yīng)零點時間氣相組分中不存在甲烷摻入,定義置換效率η為置換出的甲烷氣量VG4與參與合成甲烷氣量ΔV的比值,與式(6)和式(9)聯(lián)立求得η:
(13)
式中:η為置換效率;V為填砂孔隙體積,cm3。
2 引入多組分狀態(tài)方程改進模型
最終置換效率的精確計算需要確定甲烷的氣相密度,一般通過狀態(tài)方程計算壓縮因子后求得,而不同狀態(tài)方程計算的壓縮因子差別較大,由于水合物合成和置換過程中,壓力會發(fā)生變化,因此壓縮因子的微小偏差會帶來較大的實驗誤差。利用Visual Basic對Setzmann方程、SRK方程、RK方程、PR方程、vanderwaals方程、PRSV1方程、PRSV2方程和ESD方程進行編程并計算甲烷在300 K下0到50 MPa下的壓縮因子,見圖1。
由圖1結(jié)果可以看出來,Setzmann方程計算出的結(jié)果與NIST數(shù)據(jù)庫中壓縮因子幾乎相等。而其他的公式則在5 MPa之后產(chǎn)生較大偏差以NIST美國國家標(biāo)準(zhǔn)與技術(shù)研究院數(shù)據(jù)庫中甲烷的壓縮因子為參考,與各個方程計算出的結(jié)果進行誤差分析,見圖1~2。

圖1 300 K時各方程計算壓縮因子

圖2 300 K時各方程計算相對誤差
3 模型計算精度分析
常規(guī)關(guān)于水合物合成過程中氣相組分的計算,均采用PR方程[17-18],分析圖2數(shù)據(jù)顯示該方法計算所得壓縮因子在水合物合成壓力范圍內(nèi)較NIST數(shù)據(jù)庫中的壓縮因子存在較大誤差,現(xiàn)對改進后的新模型與PR方程精度進行分析。
李旭光[19]通過實驗分析0.9、4.0、5.2、7.0℃下水合物含氣率隨時間變化的規(guī)律,合成體系溫度越低,水合物含氣率上升越快,且含氣率峰值越大。分析李旭光實驗中數(shù)據(jù),有初始注水體積VW1=150 mL,反應(yīng)孔隙體積V=2 050 mL,初始反應(yīng)壓力p1=5.8 MPa,初始反應(yīng)溫度T1=274.05 K,同時定義體積含氣率為標(biāo)況下參與反應(yīng)的甲烷體積ΔV與水合物體積比。根據(jù)李旭光所給的溫度和壓力在前200 min隨時間變化圖,利用GetData軟件對圖形進行數(shù)值化,取橫坐標(biāo)軸網(wǎng)格步長dx=2,獲得不同時刻溫度和壓力數(shù)據(jù),對比分析PR和Setzmann氣相組分狀態(tài)方程,通過式(5)和式(6)計算求得參與反應(yīng)體積ΔV,最后求得體積含氣率,得出兩種不同狀態(tài)方程不同時刻體積含氣率與實驗值對比圖形,具體見圖3。
分析圖3數(shù)據(jù)可知,在合成反應(yīng)初始階段,即誘導(dǎo)合成階段,兩種氣相組分狀態(tài)方程計算值相差不大,解釋為該階段為水合物合成的誘導(dǎo)期,此時僅有少量的甲烷分子克服表面張力溶解在體系溶液中,此時體積含氣率較低,兩種方程計算差值不大;后期進入到水合物快速合成階段,體積含氣率迅速增大,前文分析顯示PR狀態(tài)方程計算壓縮不準(zhǔn),致使在氣體體積和體系壓力大量變化時出現(xiàn)較大誤差,反映在圖3上為PR方程較實測數(shù)據(jù)點出現(xiàn)較大誤差。
針對置換體系的效率情況,單純分析式(13),顯示置換溫度降低,置換效率升高,解釋為置換過程為放熱過程,通過降低置換溫度,置換平衡將會正向移動,增大置換效率。邢艷青等人[20]通過實驗獲得不同溫度下二氧化碳置換甲烷氣體程度圖,見圖4。

圖3 新模型計算體積含氣率與實測含氣率對比圖
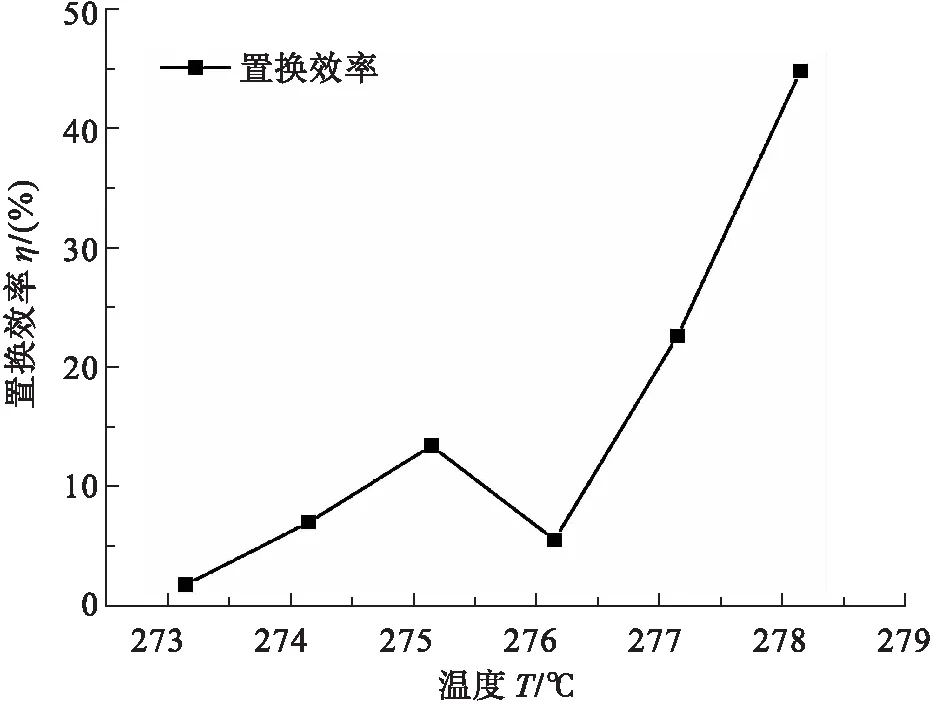
圖4 不同溫度下二氧化碳置換甲烷氣體程度圖
分析圖4可知,置換過程隨著溫度的增加,置換程度呈現(xiàn)整體上升的趨勢,解釋為實驗條件下獲得的置換效率為甲烷水合物分解量和置換開始前水合物中總的甲烷量的比值,考慮到置換過程中會有水合物的熱解,造成置換效率計算不準(zhǔn)。分析圖4置換效率在低溫條件下出現(xiàn)低值,解釋為置換初期,置換發(fā)生在甲烷水合物表面,在鎧甲效應(yīng)的作用下,形成的二氧化碳水合物會覆蓋在甲烷水合物表層,由于二氧化碳水合物具有更高的相平衡溫度,外部熱源傳導(dǎo)必須穿透二氧化碳水合物層,導(dǎo)致甲烷水合物瞬時熱解率降低,該溫度條件下置換權(quán)重大于水合物熱解,隨著溫度升高,熱解逐漸占主要部分,后期測得置換效率逐漸變大。
4 結(jié)論
1)目前,置換開采天然氣水合物主要存在置換效率低的問題,在內(nèi)部條件下通常表現(xiàn)為通過將置換反應(yīng)物二氧化碳的注入形式由氣態(tài)變?yōu)槿榛?從而在一定程度上提高置換效率;在外部條件下通過增大反應(yīng)體系的溫壓條件,實現(xiàn)水合物的熱解和二氧化碳分子更高的接觸程度,從而獲得相對較高的置換效率。考慮到壓力控制主要通過改變反應(yīng)物二氧化碳的注入量來實現(xiàn)調(diào)節(jié),置換過程中的壓力調(diào)節(jié)相對困難,同時,較溫度而言,壓力對置換反應(yīng)的影響程度較小,反映為置換過程主要通過溫度調(diào)節(jié)提高置換程度。
2)通過分析溫度對置換程度的影響情況,推導(dǎo)出置換程度隨溫度變化的關(guān)系模型,獲得不同溫度條件下置換反應(yīng)程度計算模型。考慮到置換過程中,一方面溫度升高雖然能增加甲烷氣體的產(chǎn)出,但是當(dāng)溫度升高到二氧化碳水合物的相平衡溫度之上時,將會導(dǎo)致二氧化碳水合物的二次分解;另一方面,在常規(guī)的接觸熱交換升高置換反應(yīng)體系溫度情況下,依據(jù)熱力學(xué)第三定律,當(dāng)溫差越大,熱損失越嚴(yán)重,反映為熱交換成本增加,經(jīng)濟適用性降低。通過本文推導(dǎo)的置換程度隨溫度變化模型,考慮經(jīng)濟成本條件下,合理提高置換反應(yīng)體系溫度,從而獲得較高置換效率下最優(yōu)化的置換體系溫度。
猜你喜歡
瘋狂英語·初中天地(2021年5期)2021-07-21 02:24:28
甘肅教育(2020年14期)2020-09-11 07:57:42
中學(xué)生數(shù)理化(高中版.高考數(shù)學(xué))(2020年5期)2020-06-02 09:19:08
商周刊(2017年9期)2017-08-22 02:57:49
遼寧經(jīng)濟(2017年6期)2017-07-12 09:27:16
中國衛(wèi)生(2016年9期)2016-11-12 13:27:54
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國洗滌用品工業(yè)(2015年7期)2015-02-28 19:02:38
電子設(shè)計工程(2015年12期)2015-02-27 12:06:10
中國衛(wèi)生(2014年11期)2014-11-12 13:11:32
