芽孢桿菌S6內切葡聚糖酶基因的克隆及功能鑒定
2018-06-26 04:32:30,,,
濟南大學學報(自然科學版) 2018年4期
, , ,
(濟南大學 生物科學與技術學院, 山東 濟南 250022)
目前被廣泛應用的生防菌主要為芽孢桿菌,種類有枯草芽孢桿菌(Bacillussubtilis)[1-2]、多粘芽孢桿菌(Bacilluspolymyxa)[3]、巨大芽孢桿菌(Bacillusmegaterium)、地衣芽孢桿菌(Bacilluslicheniformis)[4]、蘇云金芽孢桿菌(Bacillusthuringiensis)[5]、解淀粉芽孢桿菌(Bacillusamyloliquefaciens)[6-7]、蠟樣芽孢桿菌(Bacilluscereus)等[8],其中,生防芽孢桿菌抑制病害的機理主要為拮抗作用[9-10]。人們已從拮抗菌中提取出了多種抗菌物質, 有小分子的抗生素、蛋白性的抗菌物質、多肽抗生素以及次生代謝產生的抗菌物質等[11]。
真菌的細胞壁主要由葡聚糖和幾丁質構成, 葡聚糖酶和幾丁質酶共同作用,可以破壞真菌菌絲體頂端細胞壁,使菌絲體頂端膨脹、破碎,最終死亡[12]。根據葡聚糖酶作用于糖苷鍵位點的不同,可分為β-1, 2、β-1, 3、β-1, 4、β-1, 6-葡聚糖酶。葡聚糖水解作用是由上述多種葡聚糖酶共同作用的結果。魏艷麗[13]等從巨大芽孢桿菌AP25中克隆了內切葡聚糖酶編碼基因。本文中以自行分離篩選得到的能夠產生β-1, 4-內切葡聚糖酶基因(glu)的天然芽孢桿菌S6為出發菌株,進行glu的克隆和序列分析,并在大腸桿菌(Escherichiacoli,E.coli)中進行表達,為開發高效的拮抗新產品提供理論依據和應用基礎。
1 材料與方法
1.1 菌株和載體
菌株:BacillussubtilisS6,本實驗室自主篩選;E.coliJM110和E.coliBL-21,本實驗室保存;
載體:pGEM-T載體體系(帶有氨芐抗性),天根生化科技(北京)有限公司;pET-21b(帶有氨芐抗性),本實驗室保存。
1.2 培養基及主要試劑
大腸桿菌液體培養基: 酵母粉5 g, 胰蛋白胨10 g, 氯化鈉10 g, 定容至1 000 mL, pH=7.0, 121 ℃高壓蒸汽滅菌20 min。
羧甲基纖維素培養基:羧甲基纖維素10 g,磷酸氫二鉀 1.87 g,檸檬酸1.252 g,瓊脂粉10~15 g,定容至1 000 mL,pH=7.0,121 ℃ 高壓蒸汽滅菌20 min。
瓊脂糖凝膠電泳: 稱取0.8 g瓊脂糖, 溶于100 mL電泳緩沖液, 混勻,微波爐加熱煮沸至瓊脂糖全部溶解。
限制性內切酶、 T4脫氧核糖核酸(DNA)連接酶,寶生物工程有限公司; 聚合酶鏈式反應試劑,南京諾唯贊生物科技有限公司; 氨芐青霉素,國藥化學試劑公司; DNA分子量標記物,北京博邁德公司; 蛋白分子量標記物,北京天為時代公司; 回收試劑盒和β-半乳糖苷酶的顯色底物,Promega公司;異丙基-β-D-硫代半乳糖苷,Fermentas公司。引物合成、測序工作,由北京擎科新業生物技術有限公司完成;其余實驗所用試劑均為分析純。
1.3 芽孢桿菌S6內切葡聚糖酶基因的聚合酶鏈式反應擴增
以芽孢桿菌S6基因組為模板,根據基因序列數據庫(GenBank)登陸的β-1, 4-內切葡聚糖酶基因(登錄號為AP009484.1、 CP001982.1、 AE017355.1)同源性序列,設計聚合酶鏈式反應(PCR)引物為上游序列(S-GLU-S02):5’-ATGAAACGGTCAATCTCTAT-3’;下游序列(S-GLU-A02):5’-CTAATTTGGTTCTGTTCCCC-3’。PCR反應程序如下:94 ℃預變性10 min,94 ℃變性60 s,48 ℃退火45 s,72 ℃延伸90 s,循環次數為35;72 ℃延伸10 min。PCR產物經瓊脂糖凝膠電泳分離,使用北京博邁德瓊脂糖凝膠DNA回收試劑盒回收DNA片段。
1.4 芽孢桿菌S6內切葡聚糖酶基因序列的測定和分析
將瓊脂糖凝膠電泳回收產物與pGEM-T Easy載體連接,轉化E.coliJM110感受態細胞,篩選陽性轉化子進行測序。用SignalP3.0 Server 和TMHMM2.0在線軟件對基因序列及氨基酸序列進行序列分析。
1.5 芽孢桿菌S6內切葡聚糖酶基因原核表達
以重組質粒為模板,根據測序所得序列設計引物為上游序列(S-GLU-S03):5’-GAATTCGCATATGAAACGGTC-3’;下游序列(S-GLU-A03): 5’-CACCAAAGCTTCTAATTTGG-3’。將純化后的PCR產物和表達載體pET-21b分別用NdeI和HindIII雙酶切后進行連接與轉化入表達菌株E.coliBL21中, 挑取單菌落37 ℃培養至光密度(optical density,OD)值為0.6左右, 加入誘導劑異丙基-β-D-硫代吡喃半乳糖苷(isopropyl β-D-thiogalactoside,IPTG)至終質量濃度為100 mg/L, 37 ℃培養3 h,收集菌體、裂解,以未誘導的菌體裂解液做對照,聚丙烯酰胺凝膠電泳分析內切葡聚糖酶基因的表達。
1.6 芽孢桿菌S6內切葡聚糖酶基因功能鑒定
將轉入pET-21b-glu質粒的BL-21菌株經誘導后點接在羧甲基纖維素固體培養基,37 ℃培養約2 h;在平板中加入1 mL(質量濃度為1 g/L)的剛果紅溶液染色20 min,倒去染色液并用氫氧化鈉NaOH(濃度為1 mol/L)溶液沖洗,去掉表面附著的剛果紅,觀察是否出現透明水解圈。
2 結果與分析
2.1 芽孢桿菌S6內切葡聚糖酶基因的PCR擴增
以芽孢桿菌S6基因組DNA為模板進行PCR擴增,PCR產物經瓊脂糖凝膠電泳檢測,獲得大小約1 500 bp的條帶(見圖1),與預期的內切葡聚糖酶基因大小一致。

M為200 bp蛋白分子量標記物;1為PCR產物。圖1 聚合酶鏈式反應(PCR)擴增芽孢桿菌S6基因組上的glu基因
2.2 芽孢桿菌S6內切葡聚糖酶基因序列的測定和分析
PCR產物經過T載體克隆后驗證并測序,測序結果分析得到1 500 bp含有完整閱讀框的基因序列,經過比對分析,初步確定該序列為內切葡聚糖酶基因全長序列,并于GenBank上注冊,GenBank登錄號為HQ650233。
分別將1 500 bp閱讀框架及其編碼產物在美國國家生物技術信息中心(National Center for Biotechnology Information,NCBI)中進行核苷酸序列比對(見表1)和氨基酸序列比對(見表2)。芽孢桿菌S6內切葡聚糖酶基因與Bacilluslicheniformiscellulase內切葡聚糖酶基因具有98%相似,而與不同屬的PaenibacilluspolymyxaCel5A的僅有69%的一致性。說明內切葡聚糖酶基因在同屬菌株中序列相似性很高,進化保守;而不同屬間的相似性低,進化趨異。
2.3 芽孢桿菌S6內切葡聚糖酶二級結構分析
用SignalP3.0 Server在線軟件對芽孢桿菌S6內切葡聚糖酶氨基酸序列中的信號肽序列進行了分析, 結果如圖2所示。 用TMHMM2.0在線軟件對芽孢桿菌S6glu氨基酸序列中可能的跨膜區段進行了分析,結果如圖3所示。 芽孢桿菌S6內切葡聚糖酶氨基酸序列第1—29氨基酸具有明顯的信號肽序列信號, 蛋白的成熟位點在第29、 30的氨基酸殘基之間, 第1—30氨基酸為跨膜螺旋信號, 初步推測第1—30個氨基酸序列是芽孢桿菌S6glu的信號肽序列。
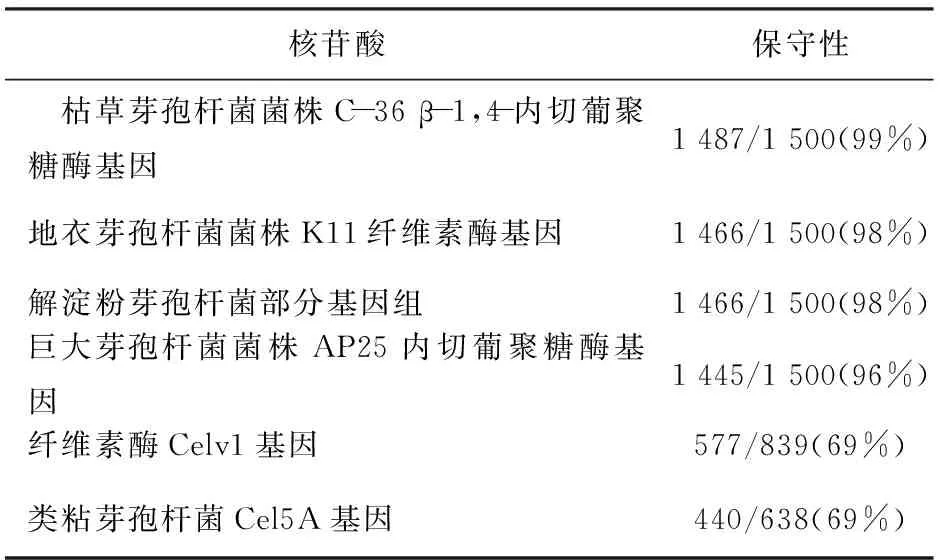
表1 芽孢桿菌S6 β-1, 4-內切葡聚糖酶基因的序列對比分析結果
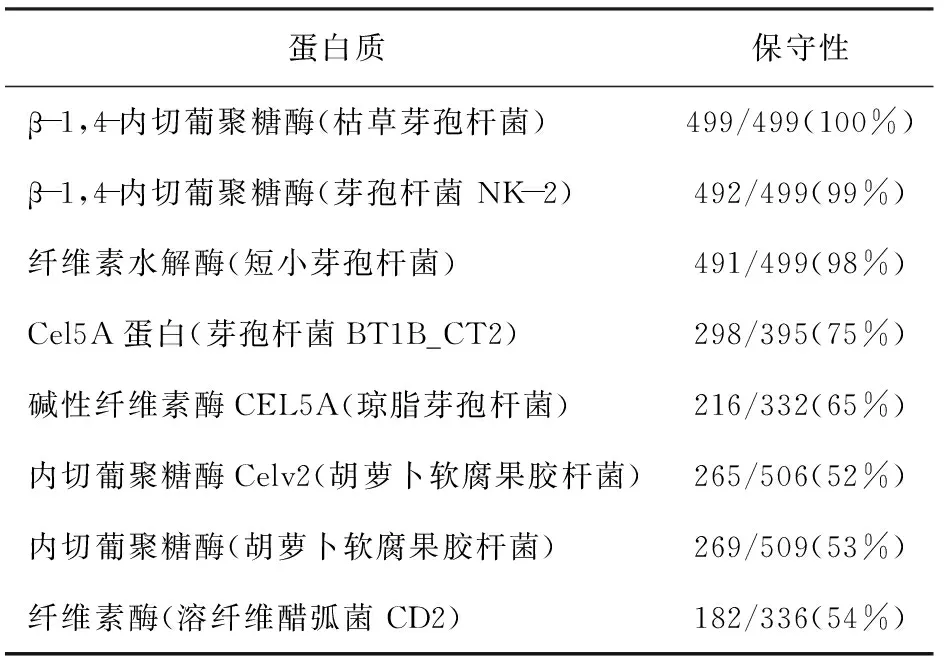
表2 芽孢桿菌S6 β-1, 4-內切葡聚糖酶的序列對比分析結果
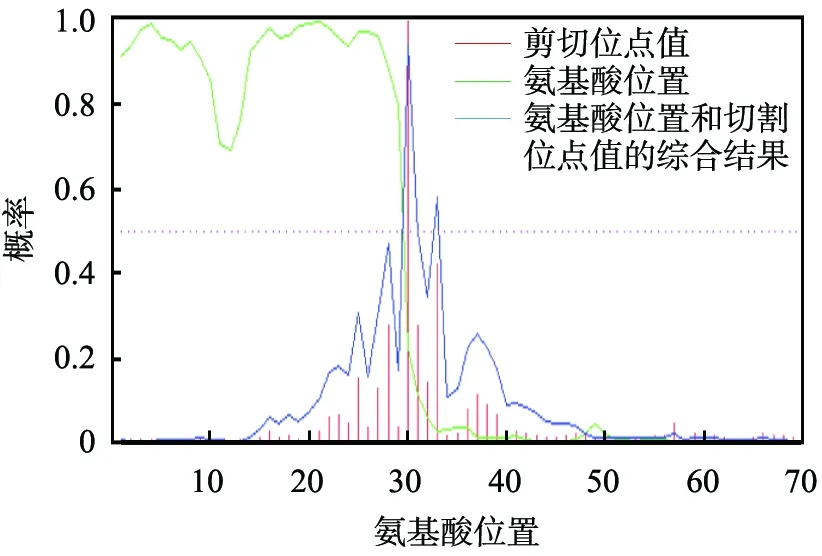
圖2 芽孢桿菌S6 glu 信號肽序列分析結果
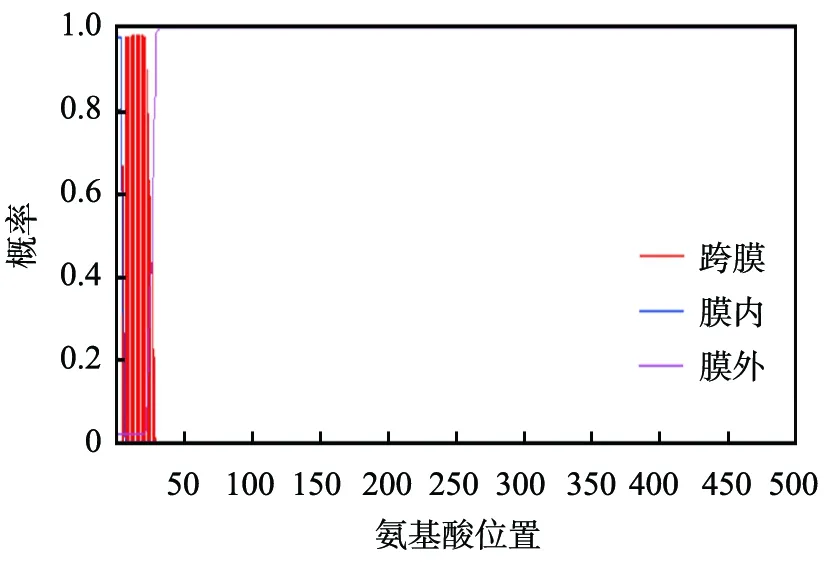
圖3 芽孢桿菌S6 glu跨膜螺旋信號分析結果
2.4 芽孢桿菌S6內切葡聚糖酶基因原核表達
將構建好的表達載體進行NdeⅠ和EcoRⅠ酶切驗證,瓊脂糖凝膠電泳檢測得到大小約為1 500 bp的條帶,與預期的酶切條帶大小相符(見圖4)。將該重組質粒進一步測序,通過測序結果得知構建完成pET-21b-glu表達載體,同時擁有正確的閱讀框。將含有pET-21b-glu重組質粒的E.coliBL21菌株,經過IPTG誘導之后,以未誘導的E.coliBL21菌株作為對照,通過聚丙烯酰氨凝膠電泳,發現在分子量約50 ku處有明顯的表達條帶,是內切葡聚糖酶基因蛋白表達的產物,對照在該位置未出現明顯的表達條帶(見圖5),表明了表達載體pET-21b-glu在E.coliBL21菌株中成功表達。
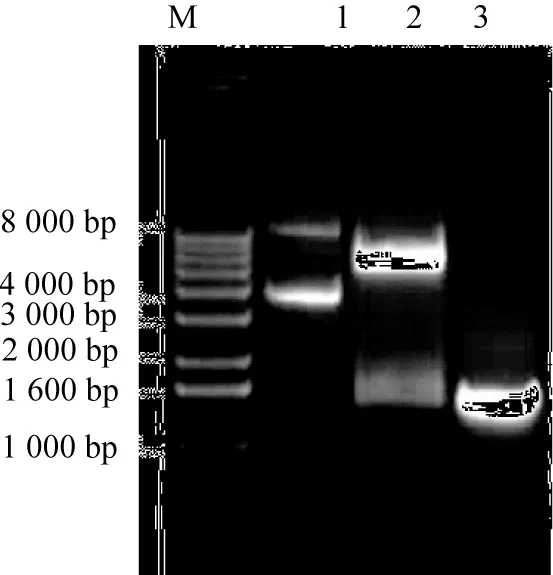
M為1 kb蛋白分子量標記物;1為pET-21b-glu質粒;2為NdeⅠ、Hind Ⅲ雙酶切質粒pET-21b-glu;3為菌落PCR。圖4 pET-21b-glu質粒雙酶切與聚合酶鏈式反應(PCR)電泳結果
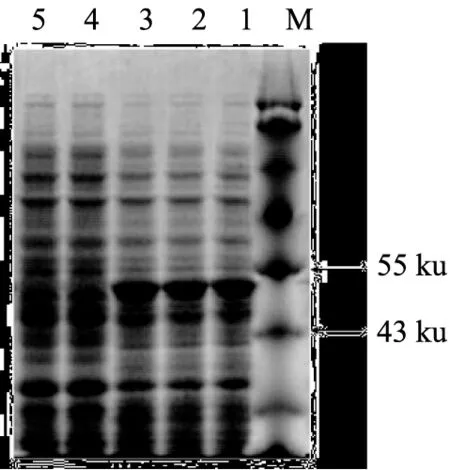
M為即時可用的蛋白質分子量標記物MW,ku;1—3為GLU誘導表達;4—5為對照。圖5 聚丙烯酰氨凝膠電泳檢測
2.5 芽孢桿菌S6內切葡聚糖酶基因功能的鑒定
將轉入pET-21b-glu質粒的BL-21菌株經誘導后點接在羧甲基纖維素固體培養基上,培養2 h,經剛果紅染色,觀察到該菌株產生明顯的內切葡聚糖酶水解圈,見圖6。
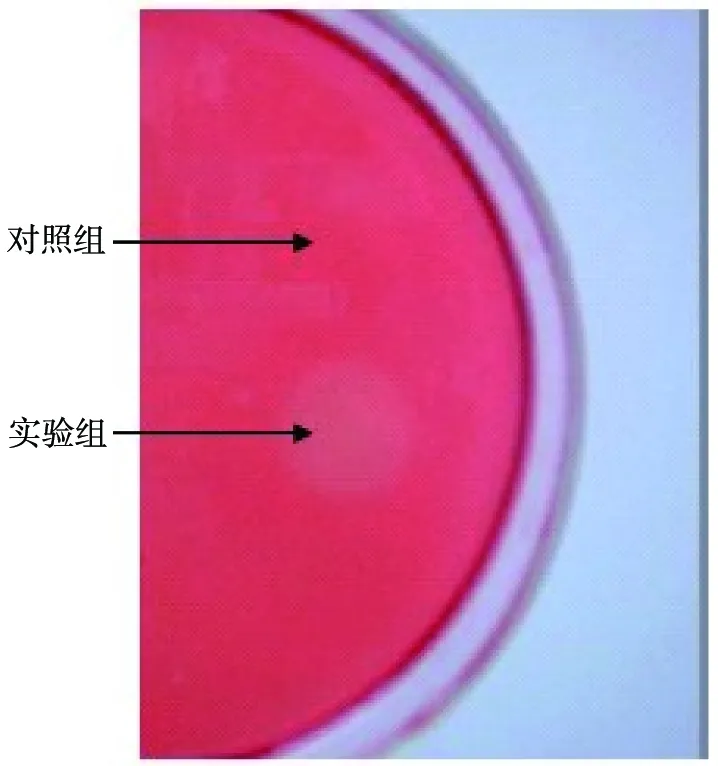
圖6 β-1, 4-內切葡聚糖酶活檢測
3 結果與討論
芽孢桿菌S6是本實驗室從采集自中國農業科學院植物保護研究所黃萎病、 枯萎病發病較輕的試驗棉田的根圍土中分離出的, 是一種對棉花黃萎病、 枯萎病病菌具有明顯拮抗作用的芽孢桿菌。 用同源克隆的方法獲得了芽孢桿菌S6的β-1, 4-內切葡聚糖酶基因。 基因序列分析該基因讀碼框為1 500 bp, 編碼499個氨基酸。陳惠等[14]將外源的內切葡聚糖酶基因轉入巨大芽孢桿菌WH320中,獲得有效表達。本文中首次從本實驗室具有自主知識產權的高效拮抗菌芽孢桿菌S6中獲得β-1, 4-內切葡聚糖酶基因。
真菌細胞壁主要由葡聚糖和幾丁質構成,根據葡聚糖酶作用于葡聚糖的位點不同,可以將葡聚糖酶分為β-1, 3-葡聚糖酶、β-1, 4-葡聚糖酶等。葡聚糖酶和幾丁質酶共同作用,可以破壞真菌菌絲體頂端細胞壁,使菌絲體頂端膨脹、破碎,最終使病原菌生長受阻或細胞死亡,阻止病害的發生和發展。Hong等[15]報道,一株類芽孢桿菌產生的β-1, 3-葡聚糖酶能破壞植物病原真菌瓜果腐霉菌和立枯絲核菌的細胞壁結構。 本文中從芽孢桿菌S6中克隆得到的內切葡聚糖酶基因為與枯草芽孢桿菌C-36的β-1, 4-葡聚糖酶的基因具有99%的相似性。 通過構建原核表達載體, 將其轉入表達宿主大腸桿菌BL-21中進行glu原核表達檢測, 發現芽孢桿菌S6的β-1, 4-葡聚糖酶基因表達產物為分子量約50 ku的蛋白。 通過對β-1, 4-內切葡聚糖酶活性進行檢測,發現芽孢桿菌S6的β-1, 4-葡聚糖酶基因在大腸桿菌中實現表達并且具有顯著的酶活性。目前,正在進行幾丁質酶基因的克隆,將幾丁質酶基因和β-1, 4-葡聚糖酶基因共同構建植物表達載體,并轉染作物,以期獲得多功能的抗性植株。
