基于詞向量的特征詞選擇
2018-06-20 07:51:06彭昀磊
計算機技術與發展 2018年6期
關鍵詞:單詞
彭昀磊,牛 耘
(南京航空航天大學 計算機科學與技術學院,江蘇 南京 210016)
0 引 言
通過相互作用,細胞中的蛋白質完成細胞中的大部分過程,比如細胞內通訊。因而,蛋白質交互信息(protein-protein interaction,PPI)成為了關鍵信息,用以解決大量醫學難題。目前,生物學家通過人工閱讀的方式識別醫學文獻中的PPI,并按照統一的格式將這些重要的信息錄入數據庫,如HPRD[1]、IntAct[2]、MINT[3]。然而以上數據庫中的PPI信息并不全面,而且生物醫學的快速發展導致每年相關科學文獻的增長數量達上千萬,每天也在產生新的蛋白質之間的關系。因此要從醫學文獻中收集PPI信息,僅靠手工方式難以滿足現實的需求。
在此背景下,有監督的機器學習方法被大量地運用到研究PPI關系識別中,但需要依賴于大量的文本集合,并要求文本集合質量高且標注蛋白質交互信息,而構造這樣的集合需要耗費大量的人力和時間。因此,筆者在之前的研究中提出了一種基于弱監督的蛋白質交互識別法,只需利用少量已有的標注信息進行蛋白質交互關系識別。該方法對蛋白質關系描述的上下文進行聚類,提取出交互關系描述的模式,用模式對交互關系進行判斷。文中在該方法的基礎上,提出了基于詞向量的特征詞選擇。為特征詞集合中的每個單詞產生一個向量,再根據單詞對應的向量將單詞聚類,然后從聚類結果中選出更能表達PPI信息的詞,繼而進行PPI識別。
1 相關工作
近年來,研究者們越來越多地采用基于機器學習的方法[4-6]去識別PPI信息。基于機器學習的方法主要包括兩大類:基于核函數的方法和基于特征的方法。基于核函數的方法首先對句子結構進行深入研究,通過設計核函數衡量不同蛋白質對間的相似度,然后使用支持核函數的分類器進行PPI關系識別。例如,Bunescu R C等[7]提出了最短依賴路徑核,以樹的形式來表示句子,用兩個實體之間的最短路徑表示實體之間的關系;文獻[8-11]使用基于圖核、多核(特征的核,樹核及圖核融合)的學習方法抽取PPI信息。基于特征的方法試圖從標注有交互關系的蛋白質對的句子中提取出對PPI識別有效的特征來建立模型,進而判斷蛋白質對是否含有交互關系。例如,文獻[12-15]從句子中的詞匯形式、位置以及蛋白質的上下文等信息中提取特征。Yang等[6,16]使用了蛋白質實體間的眾多特征,如:距離、鏈接、詞匯、關鍵詞等。但有監督的方法需要大量有標注的數據,且研究對象是單個句子,因此只能依賴一個句子中的線索,對于復雜的句子描述很難判斷。
與上述方法不同,筆者在之前的工作中采用弱監督方式,只需利用少量的標注數據,利用與交互關系的模式的相似性對交互關系進行判斷。基于該方法,文中利用基于詞向量的方法對特征詞集合中的單詞進行聚類,并從聚類結果中選出對PPI識別有效的特征詞,繼而進行蛋白質交互識別。
2 基于弱監督的蛋白質交互關系識別
基于弱監督的蛋白質交互識別主要分為四個模塊,下面簡單描述這四個模塊。
2.1 種子實例特征表示
該模塊找到文本庫中包含種子的句子,其中每個句子對應一個關系實例,然后生成關系實例的向量表示。其中每個句子的上下文都分為三個部分:第一個蛋白質左邊n個單詞(BEF)、兩蛋白質之間的單詞(BET)、第二個蛋白質右邊n個單詞(AFT)。
2.2 產生提取模式
根據PPI描述的相似性對PPI描述的上下文進行聚類,形成PPI描述的提取模式。先對每個實例賦予一個bet得分,再根據該得分,對實例從大到小排序。然后,采用單次聚類算法(single-pass clustering)對排序好的實例進行聚類,聚類生成的結果也即PPI關系的提取模式。
2.3 用提取模式尋找候選的關系實例
對待判斷關系的蛋白質對集中的每對蛋白質,和種子類似,在文本庫中掃描包含該對蛋白質的句子。對于每個句子,生成該句子對應實例的向量表示,并和2.2節產生的提取模式進行相似性比較。利用提取模式從所有句子(關系實例)中選出可能存在交互關系的實例,即候選關系實例。
2.4 用候選實例識別有交互關系的蛋白質對
每一個候選實例對應的蛋白質對,稱為候選蛋白質對。本節對候選蛋白質對打分,得分越高,該蛋白質對越有可能存在交互關系。最后,將候選蛋白質得分大于等于Ttuple(Ttuple是候選蛋白質對得分的閾值)的蛋白質對添加到種子集中,以用于下一輪的迭代,直到滿足迭代的終止條件。
3 基于詞向量的特征詞選擇
在基于弱監督的蛋白質交互關系識別中,選取特征詞的方式過于簡單,使得特征詞集合中存在大量的、不能有效表達交互關系的詞,如:show(說明)、express(表達)。
文中的目的是從特征詞集合中選出能有效表達交互關系的詞。首先,對于三個部分上下文,對每個上下文中的單詞作初步過濾(是指從特征詞集合中刪除停用詞、單字符單詞、數字、“字母+數字”的詞)。然后,從剩下的詞中過濾掉出現頻次小于等于3次的詞。接下來的處理過程主要分為四個模塊,下面詳細描述這四個模塊。
3.1 特征詞聚類
根據對眾多特征詞的觀察,發現能較好表達交互關系的詞往往很相似,能夠被劃分在同一類,如:inhabit(抑制)、induce(引起)。部分不能表達交互關系的詞也很相似,亦能被劃分在同一組內,如:cause(引起)、result(導致)。剩下的單詞和其他單詞都不相似,只能獨自歸為一類,如:member(成員)、type(類型),這些單詞一定不能表達交互關系,直接過濾掉。
3.1.1 訓練詞向量
文中描述的詞向量是distributed representation,是采用神經網絡訓練出來的向量。其基本思想是:通過訓練,將語料中的每個詞都映射成一個固定長度(N維)的向量。該詞向量克服了one-hot representation(建立一個詞表,向量維度等于詞表大小,詞表示為對應維度為1)的缺點,即容易受到維數災難(是指在涉及到向量的計算中,隨著維數的增加,計算量呈指數倍增長的現象)的困擾、不能很好地刻畫詞與詞之間的相似性。
采用的詞向量工具word2vec是Google開源的詞向量工具,其中模型采用的是Skip-Gram模型。訓練詞向量所用的語料是所有蛋白質對對應的句子。
3.1.2 將特征詞聚類并過濾僅含一個詞的類
根據單詞之間的相似性分別對bef、bet、aft上下文中的單詞進行單次聚類,得到三個聚類結果clus_bef、clus_bet、clus_aft。算法1描述了單次聚類的過程,該算法的輸入是單詞集合。
算法1:single-pass clustering。
輸入:Words={w1,w2,…,wn}
輸出:Clusters={}
1:cl1={w1}
2:Clusters={cl1}
3:forwn∈Words do
4:map={}//類號和相似性值的映射
5:forcli∈Clusters do
6:simVal=Sim(wn,cli)
7:ifsimVal>=Tsimthen
8:map=map∪{cli:simVal}
9:end if
10:end for
11:if map is not NULL then
12:sort(map,simVal)
13:index=map1[key]
14:clindex=clindex∪{wn}
15:else
16:clm={wn}
17:Clusters=Clusters∪{clm}
18:end if
19:end for
20:return Clusters
算法1的第1、2行表示將單詞集合中的第一個單詞作為第一個類中的第一個元素,第二個單詞和它作相似性比較。若相似性滿足閾值條件,則將第二個單詞加入到第一個類中,否則,創建一個新類,并將第二個單詞加入。算法1的第4~19行表示依次計算單詞wn和每一個類cli之間的相似性,選出單詞和任意類之間相似性最大的那個類clindex,并且單詞和該類滿足閾值條件,則將單詞wn添加到類clindex中。如果單詞wn和最大相似性的類都不滿足閾值條件,則創建一個新的空類,將單詞wn添加進去。算法1的第20行輸出所生成的類。
一個單詞和一個類之間的相似性是通過算法1第6行中的Sim(wn,cli)來計算的,也就是計算單詞wn和類cli內的各個成員單詞之間的相似性。對Sim(wn,cli),如果單詞wn和類cli中半數以上單詞的相似性都滿足閾值條件,那么返回最大相似性值,否則返回0。
計算兩個單詞的相似性采用如下公式:
Sim(wm,wn)=cos(wordVectorm,wordVectorn)
(1)
(2)
其中,wordVectorm和wordVectorn分別表示第m和第n個單詞對應的詞向量。
然后,在聚類結果的每個類中,對于相同詞根的詞,只留一個。如一個類中有詞bind、binds,則保留其中任意一個詞。再過濾掉僅有一個元素的類,因為基于文中的假設,這些類中的元素不能表達交互關系。
3.2 選出不能表達交互關系的高頻詞
根據觀察,可以發現出現頻數大且不能表達交互關系的詞在上下文中分布的特點是:(1)在bet上下文出現的頻數小于在bef上下文或aft上下文中出現的頻數(其中頻數可分為“存在頻數”和“總頻數”)。例如:在50個蛋白質對中,有17個蛋白質對對應句子的bef上下文含有詞“show”,且總共出現了27次,其存在頻數和總頻數分別是17和27;而只有7個蛋白質對對應句子的bet上下文含有詞“show”,且總共出現了9次,其存在頻數和總頻數分別是7和9。(2)總頻數的差值比存在頻數的差值更加明顯,即總頻數之差大于存在頻數之差。上述例子中總頻數之差為27-9=18,而存在頻數之差為17-7=10,且18>10。
因此,首先從bef、bet、aft上下文中分別選出各自的動詞。繼而統計各個動詞的存在頻數,記為x,并統計該動詞的總頻數,記為y。然后,對數據記錄按照x從大到小排序,組成三張動詞頻率表bef_v、bet_v、aft_v,表中一條數據記錄為(w,x,y)。然后,從三張動詞頻率表中選出不能表達交互關系的高頻詞。先從bef_v和bet_v中選擇高頻詞,選擇的方式如下:
(1)在bef_v和bet_v動詞頻率表中都存在;
(2)在bef_v頻率表中的排名比在bet_v中的排名更加靠前;
(3)同一個單詞,其在bef_v和bet_v中的總頻數之差(y1-y2)比存在頻數之差(x1-x2)更大。
三種方式的結果組成單詞集合wordSet1。對于aft_v和bet_v,其選擇方式與bef_v、bet_v之間的選擇方式類似,其選擇結果組成單詞集合wordSet2。
3.3 過濾含有不能表達交互關系單詞的類
由于3.1節得到的類中,每個類中的單詞相互之間都是比較相似的,因此,一旦某個類中有單詞被判定為不能表達交互關系,那么包含該單詞的類的所有元素也都不能表達交互關系,于是把這個類刪除。
對于在3.1節中聚類得到的clus_bef、clus_bet、clus_aft,以及在3.2節中得到的單詞集合wordSet1、wordSet2,從類clus_bef中去掉含有集合wordSet1中單詞的類,從類clus_aft中去掉含有集合wordSet2中單詞的類,從類clus_bet中去掉含有集合wordSet1、wordSet2中單詞的類。
3.4 過濾掉bet部分不存在的詞
在一個句子中,bet上下文比bef和aft部分上下文更有可能出現能較好表達交互關系的詞,因此,假如某些詞只在bef或aft中出現,而未在bet中出現過,那么這些詞不能表達交互關系,應該被過濾掉。
在剩余的類clus_bef、clus_bet、clus_aft中,對于類中的每個單詞分別求詞根,并從bef部分中去掉bef部分存在而bet部分不存在的詞根,從aft部分中去掉aft部分存在而bet部分不存在的詞根。
4 實 驗
4.1 實驗數據
實驗中有交互關系的蛋白質對是直接從HPRD數據庫中查詢獲取,并且只保留被PubMed數據庫中一篇以上摘要包含的那些蛋白質對。而對于無交互關系的蛋白質對,將HPRD中的蛋白質隨機組合成蛋白質對,去除已被HPRD數據庫包含的蛋白質對以及未被PubMed數據庫記載的蛋白質對。以一對蛋白質為查詢參數,從文獻中檢索出描述這兩個蛋白質的所有句子,作為該蛋白質對的簽名檔。
文中設置一個句子中BET上下文的有效單詞個數為6(有效單詞個數是指不包括標點在內的單詞個數,不夠6個則取BET中所有的單詞)。滿足此要求的有交互和無交互關系的蛋白質對分別有964和870對,總共1 834對。這些蛋白質對對應的簽名檔構成文本庫。從有交互關系的蛋白質對中隨機挑選出50對作為種子。
4.2 實驗設置
訓練詞向量時有以下參數:min_count指最低頻率,若一個詞在預料中出現次數小于最低頻率,就放棄該詞;size指每個詞的向量維度;window指訓練時的上下文掃描窗口大小,如window為2就是考慮前2個詞和后2個詞。設置min_count為1,size為200,window為5。
使用精度(P)、召回率(R)和F值(F)作為評價標準,它們的計算公式為:
(3)
(4)
(5)
為設定算法1中的單詞相似性閾值wTsim,將wTsim從[0,1]變化,以0.1為間隔,觀察聚類結果。當類內單詞越相似,類間單詞越迥異,聚類結果越理想,最終確定wTsim為0.5。在基于弱監督的蛋白質交互識別中,為取得合理的實例相似性閾值iTsim,將iTsim從[0.1,0.6]變化(避免F值過低),以0.1為間隔,記錄實驗結果。
4.3 實驗結果及分析
表1列舉了采用特征詞選擇前后,每個實例對應bef、bet、aft部分的特征詞數量。
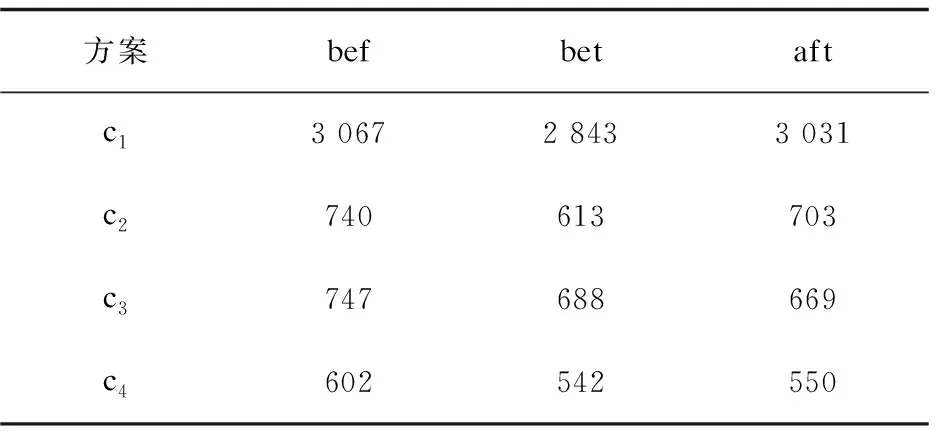
表1 各方案下的特征詞數量
表1中,c1表示未采用特征詞選擇;c2表示用詞向量進行聚類后,過濾僅含一個元素的類;c3表示未使用詞向量進行聚類,只過濾掉不能表達交互關系的高頻詞以及bet中不存在的詞;c4表示用詞向量進行聚類,過濾僅含一個元素的類,根據不能表達交互關系的高頻詞,從聚類結果中過濾掉含有高頻詞的類,再過濾掉bet中不存在的詞。從c1到c4,三個上下文的特征詞數量大量減少。
分別采用方案c1~c4,得到不同實例相似性閾值iTim下的F值,如圖1所示。從圖1可以看出,方案c2和c3的F值基本比c1更優,表明特征詞選擇后識別結果更優。
分別采用c2和c3,得到不同實例相似性閾值iTim下的識別結果,如表2所示。
從表2可以發現,隨著實例相似性閾值iTim減小,方案2和3的識別精度下降,召回率上升,F值上升。所有閾值iTim下,方案c3的精度都比方案c2的高。當閾值iTim在0.2及以下時,方案3的召回率和F值都比方案2的低;當閾值iTim在0.3及以上時,方案3的召回率和F值都比方案2的高。
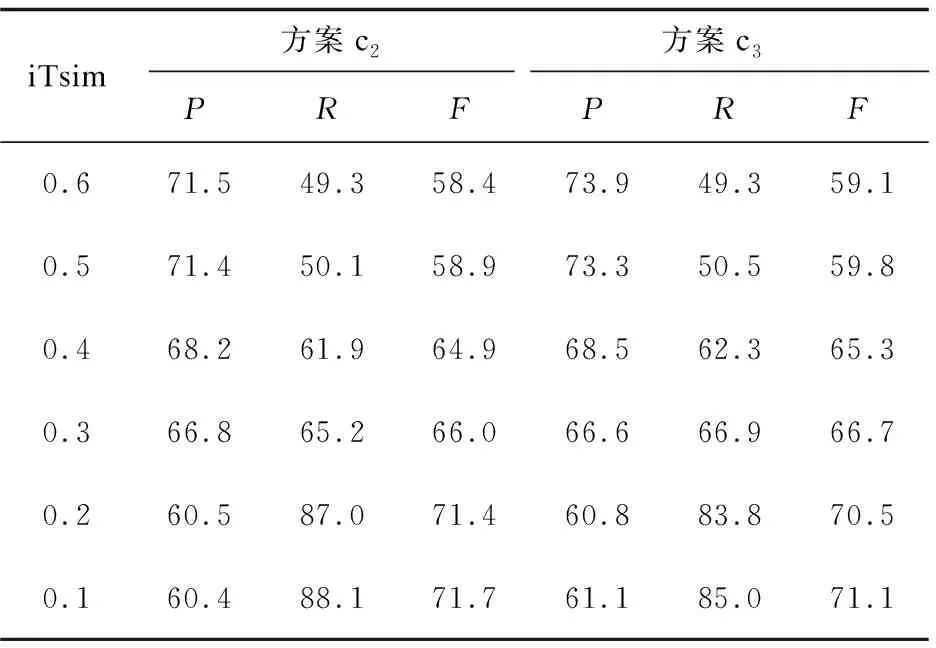
表2 采用方案c2和c3的識別結果 %
文中使用多種特征詞選擇方法,使得特征詞數量大幅減小,實例對應的特征向量維數明顯降低,極大提高了蛋白質交互的識別效率,且識別結果更優。
5 結束語
為了從特征詞中選擇表達交互關系的詞,提出了基于詞向量的特征詞選擇方法。該方法在比較單詞相似性時,使用了詞向量的方式,取得了較好的識別結果。但在選擇不能表達交互關系的高頻詞時,僅考慮其在上文中的分布情況,下一步的研究將嘗試其他的選擇方法。
參考文獻:
[1] PRASAD T S K,GOEL R,KANDASAMY K,et al.Human protein reference database-2009 update[J].Nucleic Acids Research,2009,37:767-772.
[2] KERRIEN S,ALAM-FARUQUE Y,ARANDA B,et al.IntAct-open source resource for molecular interaction data[J].Nucleic Acids Research,2007,35:561-565.
[3] CEOL A,ARYAMONTRI A C,LICATA L,et al.MINT,the molecular interaction database:2009 update[J].Nucleic Acids Research,2010,38:532-539.
[4] 崔寶今,林鴻飛,張 霄.基于半監督學習的蛋白質關系抽取研究[J].山東大學學報:工學版,2009,39(3):16-21.
[5] 董美豪.基于文本挖掘的蛋白質相互作用對抽取方法的研究[D].哈爾濱:哈爾濱工業大學,2015.
[6] 楊志豪,洪 莉,林鴻飛,等.基于支持向量機的生物醫學文獻蛋白質關系抽取[J].智能系統學報,2008,3(4):361-369.
[7] BUNESCU R C,MOONEY R J.A shortest path dependency kernel for relation extraction[C]//Proceedings of the conference on human language technology and empirical methods in natural language processing.Vancouver,British Columbia,Canada:Association for Computational Linguistics,2005:724-731.
[8] HIDO S,KASHIMA H.A linear-time graph kernel[C]//Ninth IEEE international conference on data mining.[s.l.]:IEEE,2009:179-188.
[9] AIROLA A, PYYSALO S, PAHIKKALA T, et al.A graph kernel for protein-protein interaction extraction[C]//Proceedings of workshop on current trends in biomedical natural language processing.[s.l.]:Association for Computational Linguistics,2008:1-9.
[10] 唐 楠,楊志豪,林鴻飛,等.基于多核學習的醫學文獻蛋白質關系抽取[J].計算機工程,2011,37(10):184-186.
[11] 劉 念,馬長林,張 勇,等.基于樹核的蛋白質相互作用關系提取的研究[J].華中科技大學學報:自然科學版,2013,41:232-236.
[12] NIU Y,OTASEK D,JURISICA I.Evaluation of linguistic features useful in extraction of interactions from PubMed;application to annotating known, high-throughput and predicted interactions in I2D[J].Bioinformatics,2010,26(1):111-119.
[13] 高 飛.基于MapReduce的蛋白質相互作用信息抽取系統的設計與實現[D].西安:西北農林科技大學,2016.
[14] 吳紅梅,牛 耘.基于特征加權的蛋白質交互識別[J].計算機技術與發展,2016,26(2):114-117.
[15] 張 景,吳紅梅,牛 耘.基于Minimum Cuts的蛋白質交互識別[J].計算機技術與發展,2017,27(6):17-21.
[16] 劉敏捷.基于組合學習和主動學習的蛋白質關系抽取[D].大連:大連理工大學,2015.
猜你喜歡
閱讀(快樂英語中年級)(2024年9期)2024-10-23 00:00:00
時代英語·高三(2024年3期)2024-09-03 00:00:00
時代英語·高三(2024年3期)2024-09-03 00:00:00
時代英語·高三(2024年3期)2024-09-03 00:00:00
時代英語·高三(2024年3期)2024-09-03 00:00:00
時代英語·高三(2024年3期)2024-09-03 00:00:00
時代英語·高三(2024年3期)2024-09-03 00:00:00
閱讀(快樂英語高年級)(2020年8期)2020-01-08 02:21:16
智慧少年·故事叮當(2018年11期)2018-05-14 11:48:18
意林(繪英語)(2017年5期)2017-05-15 02:17:23
