溶劑熱法合成Fe-CeO2與N-Fe-CeO2納米粉體及其光催化性能
2018-05-05 06:22:34黃建平折小梅石惠民
無機化學學報 2018年5期
黃建平 陳 芳 折小梅 王 赫 石惠民
(1湖南大學,材料科學與工程學院,長沙 410082)
(2湖南大學,物理與電子科學學院,長沙 410082)
0 引 言
近年來,隨著工業技術的發展,人民生活水平逐步提高,但環境污染問題也日益嚴峻。光催化技術是利用光能治理有機污染物、無機污染物和制取氫等的一門新興技術,引起了學者的廣泛關注。目前,除了技術相對成熟并且已獲廣泛應用的TiO2外,新型光催化劑也不斷被開發出來。其中,CeO2由于具有良好的催化活性和穩定性,較高的氧化還原性能(Ce3+/Ce4+),且成本低、無毒等特點,在光催化領域的應用前景已倍受關注[1-3]。與TiO2的寬禁帶,太陽能利用率低,量子效率低等缺點類似[4],CeO2的禁帶寬度約3.2 eV[5],通常只吸收400 nm以下的紫外光,而不能充分利用太陽光中能量集中的可見光。因此唯有降低CeO2的帶隙寬度,使其光響應范圍延伸至可見光區才能從根本上提高CeO2光催化活性。摻雜是提高CeO2光催化活性的有效途徑之一,通過適當的摻雜,在CeO2帶隙中引入雜質能級以減小其帶隙寬度。目前通過摻雜改善CeO2光催化性能的報道可分為2種:一種是摻雜過渡金屬和稀土元素,如 Fe、Co、Y、La 等[5-12]。Wang 等[7]采用水熱法、共沉淀法和溶劑熱法3種方法合成了Fe摻雜CeO2納米片,發現在100~175℃的水熱合成的Fe摻雜CeO2對二氯乙烷的光催化性能最好,而175~350℃高溫合成條件下,溶劑熱法制備Fe摻雜CeO2催化性能更佳。袁強等[8]采用溶膠-凝膠法和浸漬法制備了Fe/CeO2固體催化劑,用來催化剛果紅,其最高催化效率達96%。Arul等[9]采用溶劑熱法合成了Fe摻雜CeO2,證實了其對亞甲基藍的降解率得到提高,但并未指出Fe的最佳摻雜量。他們還合成了Co摻雜CeO2,結果證明Co-CeO2對偶氮染料的光催化性能良好[10]。Liyanage等[5]在CeO2中摻雜不同含量的Y,發現隨著Y含量的增加,對靛藍胭脂紅、羅丹明B的光催化降解性能都逐步提高。另一種是摻雜非金屬元素,如N、C等[13-19]。顧明杰等[14]以三乙醇胺為氮源,通過溶劑熱法成功制備氮摻雜納米CeO2,并且發現氮摻雜CeO2對酸性橙的降解率高達93.9%。Mao等[15]以乙二胺為氮源,通過蒸餾回流法成功將氮摻入CeO2,通過可見光催化分解亞甲基藍溶液,發現CeO2摻雜4%氮時光催化活性最好。Wu等[16]以濃硝酸為氮源,采用溶劑熱法將氮摻入CeO2,結果顯示摻氮CeO2對羅丹明B的降解率是純CeO2的12.6倍。翦立新等[19]利用微波等離子技術結合溶膠凝膠法制備了Fe3+-N-CeO2,發現當摻Fe量為0.1%時,Fe3+-N-TiO2具有最好的催化活性,而N摻雜進一步增強了對可見光的利用率。因此,通過適當的方式,摻雜適量的過渡金屬、稀土金屬或非金屬元素,在一定程度上可提高CeO2的光催化性能。但由于金屬元素與非金屬元素在CeO2晶格中摻雜位置不相同,對光催化性能的提高機制也不相同。
因此,為了考察金屬與非金屬元素共同摻雜對CeO2光催化性能的協同效果,本文以Ce(NO3)3·6H2O為鈰源,Fe(NO3)3·9H2O為鐵源,制備了不同含量Fe摻雜CeO2(Fe-CeO2),并用不同含氮溶劑為氮源,制備了不同氮源的N-10%Fe-CeO2納米粉體,并考察了其在模擬太陽光照條件下對有機污染物亞甲基藍(MB)的光催化降解性能。
1 實驗部分
1.1 主要試劑
所用的化學試劑主要有六水合硝酸鈰 (威海佰德信新材料有限公司,分析純,>98%),九水合硝酸鐵(上海阿拉丁生化科技股份有限公司,分析純,>98%),乙二醇(國藥集團化學有限公司,分析純,>99%),濃氨水(成都市科龍化工試劑廠,分析純,24%~26%),尿素(天津市恒興化學試劑制造有限公司,分析純,>99%),三乙醇胺(上海阿拉丁生化科技股份有限公司,分析純,>98%),二乙醇胺(上海阿拉丁生化科技股份有限公司,分析純,>99%),亞甲基藍(天津市恒興化學試劑制造有限公司,指示劑,>98%)。氫氧化鈉(天津市進豐化工有限公司,分析純,>98%)。
1.2 樣品的制備
1.2.1 Fe-CeO2催化劑的制備
以Ce(NO3)3·6H2O為鈰源,乙二醇為溶劑,采用溶劑熱法制備了不同摻雜含量的Fe-CeO2納米粉體。首先按照總物質的量為0.01 mol,且nFe/(nFe+nCe)=0%,5%,10%,15%分別計算出 Ce(NO3)3·6H2O 和Fe(NO3)3·9H2O 的質量。 先將 Ce(NO3)3·6H2O 溶解于35 mL乙二醇攪拌至溶解后,再加入對應的Fe(NO3)3·9H2O,再攪拌30 min至充分溶解,再加入5%NaOH溶液,調節其pH值為9~10,繼續攪拌30 min。將溶液轉移至50 mL聚四氟乙烯內襯反應釜中,在180℃的馬弗爐中反應24 h。然后隨爐冷卻至室溫,取出生成的沉淀,用去離子水和乙醇溶液反復洗滌數遍,離心分離后放入70℃干燥箱中干燥。最后將干燥好的粉體500℃煅燒2 h,即得到不同物質的量比的Fe-CeO2納米粉體。
1.2.2 N-Fe-CeO2催化劑的制備
以 Ce(NO3)3·6H2O 為鈰源,Fe(NO3)3·9H2O 為鐵源,三乙醇胺(TEA),氨水,尿素(urea),二乙醇胺(DEA)為氮源,按上述nFe/(nFe+nCe)=10%制備出N-10%Fe-CeO2粉體。首先稱取4份3.9 g的Ce(NO3)3·6H2O溶于30 mL乙二醇中,再分別加入0.404 g的Fe(NO3)3·9H2O,攪拌30 min至完全溶解混合,分別加入10.7 mL的三乙醇胺,12 mL濃氨水,4.8 g尿素,7.7 mL二乙醇胺,繼續攪拌30 min使其充分混合,將溶液轉移至50 mL聚四氟乙烯內襯反應釜中,在180℃的馬弗爐中反應24 h。樣品的洗滌、干燥與煅燒過程與Fe-CeO2粉體一致。樣品分別標記為 TEA-N-10%Fe-CeO2,NH3·H2O-N-10%Fe-CeO2,Urea-N-10%Fe-CeO2,DEA-N-10%Fe-CeO2。
1.3 樣品的表征
采用D/Max2500型X射線衍射儀(XRD)分析了樣品的物相成分,管電壓為40 kV,電流為10~450 mA, 靶材為銅靶(λ=0.154 nm), 掃描范圍為20°~80°;采用JEM-2100型透射電子顯微鏡(TEM)觀察了樣品的微觀形貌,加速電壓200 kV;采用ORIUS SC1000能量散射X射線光譜分析儀(EDS)分析了樣品的元素分布狀態。采用ESCALAB 250Xi型X射線光電子能譜(XPS)對樣品的表面化學狀態進行了分析,X射線光源為200 W單色Al Kα射線;采用LabRAM-010激光拉曼光譜儀對樣品進行拉曼分析,激發波長為532 nm;采用Agilent Cary 300紫外-可見分光光度計(UV-Vis)測試固體樣品的吸光度。
1.4 粉體的光催化性能及循環使用性能測試
取50 mg光催化劑分散于50 mL濃度為10 mg·L-1的亞甲基藍(MB)溶液,先在攪拌作用下暗反應30 min,以達到吸附脫附平衡。然后將溶液置于氙燈(PLS-Microsolar 300 W)下照射,持續不斷電磁攪拌,光源距溶液表面10 cm,光功率密度約為2 000 mW·cm-2。每隔30 min取樣,離心分離得上清液,用TU-1901紫外可見分光儀測試溶液在最大吸收波長664 nm的吸光度。由于吸光度與溶液濃度成正比,遵循朗伯-比爾定律,因此可用吸光度A/A0來表示溶液的濃度變化C/C0,從而評價光催化劑的催化效應。A,C分別為反應時間為t時的吸光度值和溶液濃度,A0,C0分別為暗反應前的吸光度值和溶液濃度。
將每次光催化試驗后的催化劑,離心分離進行回收,使用去離子水反復洗滌多次,烘干后再次測試其光催化性能。如此多次循環測試,以驗證光催化劑在循環使用時的催化性能的穩定性。
2 結果與討論
2.1 TEM分析
圖 1(a~d) 分別為純 CeO2、10%Fe-CeO2、NH3·H2O-N-10%Fe-CeO2和Urea-N-10%Fe-CeO2的TEM圖片。從圖中可以看出當添加NaOH為沉淀劑,無氮源時,所得納米顆粒具有較強的團聚性。而濃氨水為氮源時,所獲粉體分散較好,許多粉體堆積形成中空薄壁泡沫狀,而以尿素為氮源時,所獲粉體表現為無序的草葉狀。CeO2基材料的形貌對生長環境非常敏感,目前已合成了大量形貌各異的CeO2粉體[20]。本實驗中摻雜CeO2形貌主要受pH值變化,氮源的特性及與溶劑的共同作用。根據其高分辨插圖,測量了其晶面間距為0.306~0.315 nm之間,對應于螢石結構CeO2的(111)面。說明納米顆粒的表面主要是以密排面(111)面為主。對NH3·H2ON-10%Fe-CeO2粉體進行了EDS能譜分析,發現N、Fe在粉體中的分布比較均勻,也證明了N、Fe在粉體中均勻摻雜。圖1(e~h)分別為圖1(c)中框內區域的Ce、Fe、N和O等元素的分布圖。可以看出這些元素在粉體中均已存在且分布比較均勻。說明Fe、N等元素已均勻摻雜進入了CeO2晶格中。
2.2 XRD分析
利用X射線衍射儀對摻雜Fe、N-Fe的樣品的晶體結構進行了分析,如圖2所示。圖2(a)為不同含量Fe摻雜CeO2的XRD圖。圖中的衍射峰均表現為CeO2的立方螢石結構的特征峰 (PDF No.34-0394)。圖中沒有其它特征峰,表明摻雜的Fe已全部進入CeO2晶格。隨著摻雜Fe含量的增加,衍射峰的位置向高衍射角方向逐漸移動,說明摻雜使得其晶格常數減小。圖2(b)為10%Fe-CeO2中加入了不同氮源合成的樣品的XRD圖,其衍射峰同樣均表現為CeO2螢石結構的特征峰。衍射峰的位置,半高寬等外形特征基本相似。利用Scherrer公式和(111)、(200)、(220)、(311)的衍射峰的參數計算了晶粒尺寸,即:


圖 1 (a)純 CeO2、(b)10%Fe-CeO2、(c)NH3·H2O-N-10%Fe-CeO2 和(d)Urea-N-10%Fe-CeO2 的 TEM 圖;(e~h)分別為圖 1(c)中紅框內的 Ce、Fe、N、O 的元素分布圖Fig.1 TEM images of(a)pure CeO2,(b)10%Fe-CeO2,(c)NH3·H2O-N-10%Fe-CeO2and(d)Urea-N-10%Fe-CeO2;(e~h)Elements distribution of Ce,Fe,N and O for the area in frame in Fig.1(c)
式中,k為謝樂常數,取值0.89;λ為入射X射線波長,取值為0.154 nm;θ為衍射角;B為實測樣品衍射峰半高寬度。利用布拉格衍射定律和面心立方結構晶面間距的計算公式計算了晶體的晶格常數,如表1所示。晶格常數隨著摻雜Fe含量的增加不斷減小,主要是由于半徑較小的Fe3+(65 pm)取代了半徑較大的Ce4+(97 pm)的位置。而不同的氮源對其氧化鈰的晶格常數影響較小,主要是由于N3-(146 pm)離子半徑與O2-(140 pm)半徑接近。晶粒尺寸的變化不大,為10~12 nm之間。說明這種合成方法對晶粒尺寸分布的可控性強。
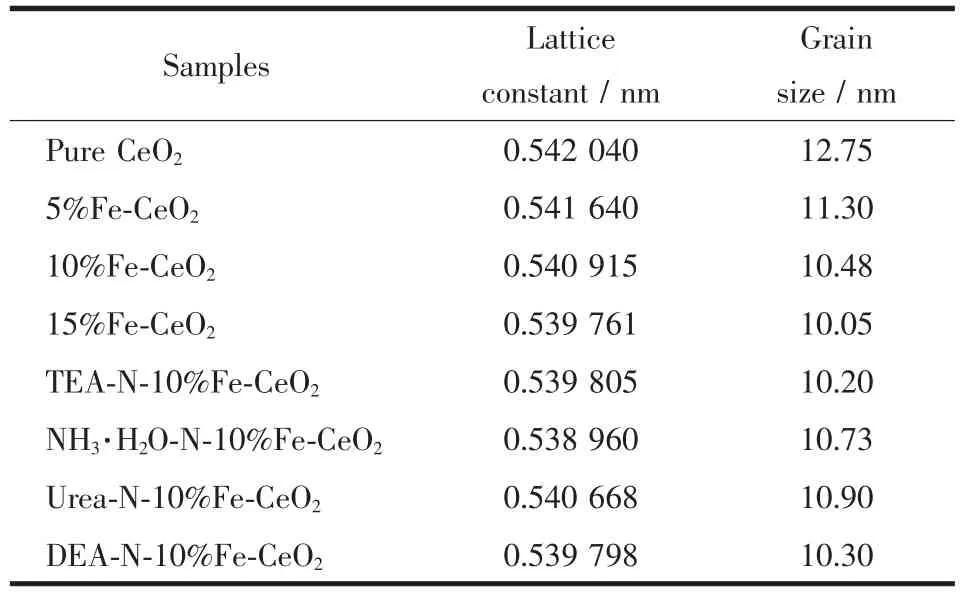
表1 樣品的晶格常數與晶粒尺寸Table 1 Lattice constants and grain sizes of the samples
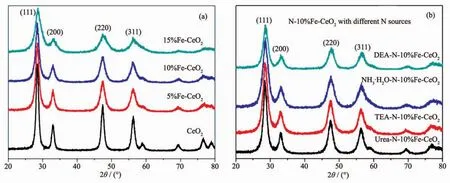
圖 2 Fe-CeO2(a)與 N-10%Fe-CeO2(b)的 XRD 圖Fig.2 XRD patterns of Fe-CeO2(a)and N-10%Fe-CeO2(b)
2.3 XPS分析
圖 3 為 NH3·H2O-N-10%Fe-CeO2與 10%Fe-CeO2中Fe2p、Fe3p和N1s的XPS高分辨圖譜。采用軟件XPSpeak4.1對Fe2p XPS圖譜進行分峰處理,如圖3(a)所示。711.4和717.4 eV分別為Fe2p3/2的主峰及衛星峰;724.4和733.2 eV分別為和Fe2p1/2是結合能主峰及衛星峰。衛星峰的強度比主峰強度高說明Fe進入CeO2晶格后,Fe與Ce之間存在著很強的相互作用。圖3(b)顯示了Fe3p的位置約為 55.6 eV,主要對應為 Fe3+[21]。 圖 3(c)中 399.2和402.9 eV分別代表N在CeO2晶格中和吸附的N2的結合能峰[18-19]。通過對比,容易看出,濃氨水摻雜后,CeO2晶格中成功摻雜了一定量的N。根據各元素結合能峰可估算出10%Fe-CeO2中nFe/(nFe+nCe)=9.2%,NH3·H2O-N-10%Fe-CeO2中 nFe/(nFe+nCe)=5.9%,nN/(nO+nN)=0.9%。
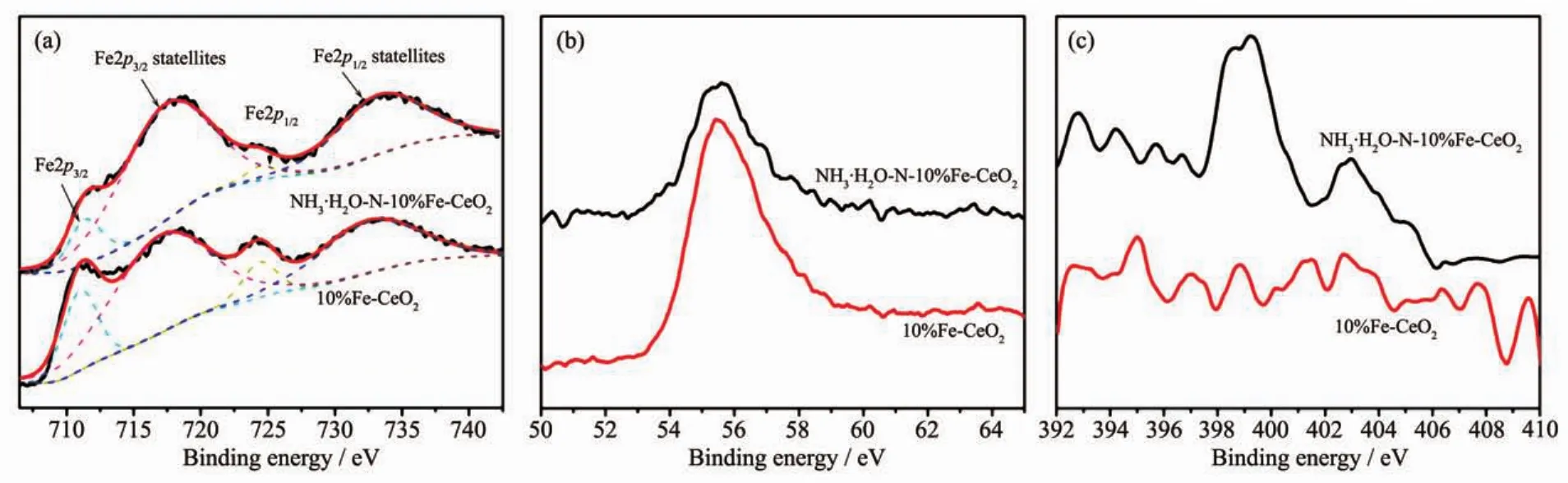
圖 3 NH3·H2O-N-10%Fe-CeO2及 10%Fe-CeO2的 Fe2p(a)、Fe3p(b)和 N1s(c)的 XPS 圖譜Fig.3 Fe2p(a),Fe3p(b)and N1s(c)XPS spectra of NH3·H2O-N-10%Fe-CeO2and 10%Fe-CeO2
2.4 Raman分析
圖4 顯示了不同含量Fe摻雜CeO2與不同氮源的N-10%Fe-CeO2的Raman圖譜。純CeO2的Raman圖譜在462.4 cm-1處存在一個F2g模式振動峰。隨著摻雜Fe含量的增加,F2g峰的位置稍有紅移,強度也逐漸降低,隨著摻雜Fe含量的增加,極化率逐漸降低,以致當Fe摻雜含量達到15%時,其F2g近乎消失。N摻雜也對F2g峰的位置和強度也有明顯的影響,如圖4(b)所示。這說明Fe、N的摻雜改變了CeO2晶格中Ce-O鍵的鍵能和原子間距,導致電子云發生了遷移。同時,Fe、N的摻雜也對晶格的極化率發生變化。另外,Raman圖譜在550~600 cm-1之間還存在一個比較寬的峰,此峰的形成反應了晶體中的氧空位濃度的變化[22-23]。
2.5 紫外-可見光譜分析
圖5(a,b)分別為不同物質的量分數的Fe摻雜CeO2和不同氮源N-10%Fe-CeO2的紫外-可見光漫反射光譜圖。可見純CeO2的吸收帶邊在400 nm附近垂直陡峭,而摻雜Fe以后,吸收帶發生明顯紅移,吸收帶邊擴展到可見光波段,緩慢降低。N的摻雜對10%Fe-CeO2有不同程度的影響,以濃氨水做氮源時擴展最大。利用半導體材料帶隙計算公式:

圖4 (a)Fe-CeO2與(b)N-10%Fe-CeO2的Raman圖譜Fig.4 Raman spectra of(a)Fe-CeO2and(b)N-10%Fe-CeO2
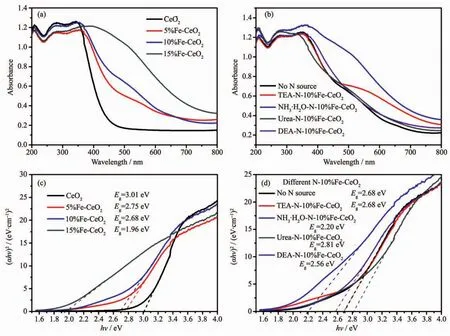
圖5 不同Fe含量的Fe-CeO2(a)與不同氮源N-10%Fe-CeO2(b)的UV-Vis吸收圖及(αhν)2~h關系圖(c,d)Fig.5 UV-visible spectroscopy of Fe-CeO2(a)and N-10%Fe-CeO2(b)and their(αhν)2~h plots(c,d)

式中,α為吸光系數;h為普朗克常數;ν為光的頻率;A為半導體材料的常數;Eg為半導體的禁帶寬度;n為常數,通常材料帶寬為直接型的n取1,間接型的n取4。氧化鈰屬于直接型帶寬材料,因此n取 1。作(αhν)2~hν關系曲線,如圖 5(c,d)所示。 曲線的切線與橫坐標的截距即為該材料的帶隙寬度Eg。從圖中可見,隨著摻雜Fe含量的增加,帶隙寬度逐漸變小。據文獻[24]報道,Fe摻雜使得導帶寬度增加,導帶底下移,并且在導帶底附近形成了一個局域能級,在原來的禁帶中也產生一個雜質能級,從而導致了禁帶寬度變窄。而N的摻雜對10%Fe-CeO2的能帶結構也有一定的微調作用,主要影響了其對可見光的吸收(圖5(b))。濃氨水做N源時帶隙寬度降低至2.20 eV,而尿素做氮源時,帶隙寬度反而升高至2.81 eV。這說明不同的氮源摻雜對晶體的電子結構起到非常重要的影響。N摻雜進入CeO2晶格中,取代了O的位置。N比O更容易失去電子,因此降低了導帶能級的高度。N的2p電子層與周圍O的2p電子層形成雜化,在價帶頂部產生新的雜質電子軌道,縮小了禁帶寬度,從而促進了光生電子與空穴的產生。
2.6 光催化性能分析
圖6為亞甲基藍溶液在氙燈的照射下的光催化降解濃度變化曲線。可見在Fe-CeO2樣品中,以10%Fe-CeO2的樣品催化效率最高,摻雜Fe含量增加到15%后,催化速度反而有所減弱。但最終Fe-CeO2的降解率都在95%左右。而純CeO2在同樣條件下對亞甲藍的降解率只有67%。摻雜N以后,10%Fe-CeO2的催化速率和催化效果產生了不同的變化。以濃氨水和二乙醇胺做氮源時,催化速度進一步提高,最終降解率達到97%。而尿素做氮源時反而使得其催化效果有所降低。這顯然與N摻雜后,其禁帶寬度的改變密切相關。禁帶寬度越窄,可吸收的紫外線和可見光越多,產生的光生電子-空穴對越多,催化降解亞甲基藍速度越快,最終降解率也越高。由于Fe3+具有未填滿的d層電子軌道,容易捕獲光電子成為Fe2+,也可捕獲空穴成為Fe4+。因此當Fe摻雜含量較低時,可成為光生電子或空穴的陷阱,促進光生載流子的分離,從而起到提高催化性能的作用。然而當Fe摻雜含量過高,Fe可能會成為光生電子與空穴的復合中心,雖然禁帶寬度變得更小,但其催化速率反而會降低[18-19]。氮摻雜后,N2p軌道與O2p軌道在導帶底部形成雜化軌道,產生雜質能級,降低了導帶能級高度,同時也在價帶頂部形成局部能級[17]。但由于摻雜的氮源不同,N的摻雜含量和摻雜狀態不同導致活性點數量和活性點的催化能力不一樣,導致其催化性能也有所不同。摻雜Fe和N對CeO2電子結構的改變和相應的催化機理可用圖7表示。不同的摻雜效應對10%Fe-CeO2的能帶結構產生了不同的影響,從而影響了其最終的催化效果。Fe與N摻雜逐漸降低了導帶的高度,并且在導帶底或價帶頂部形成局域能級,或在帶隙中形成雜質能級,從而促進了光生電荷的產生。
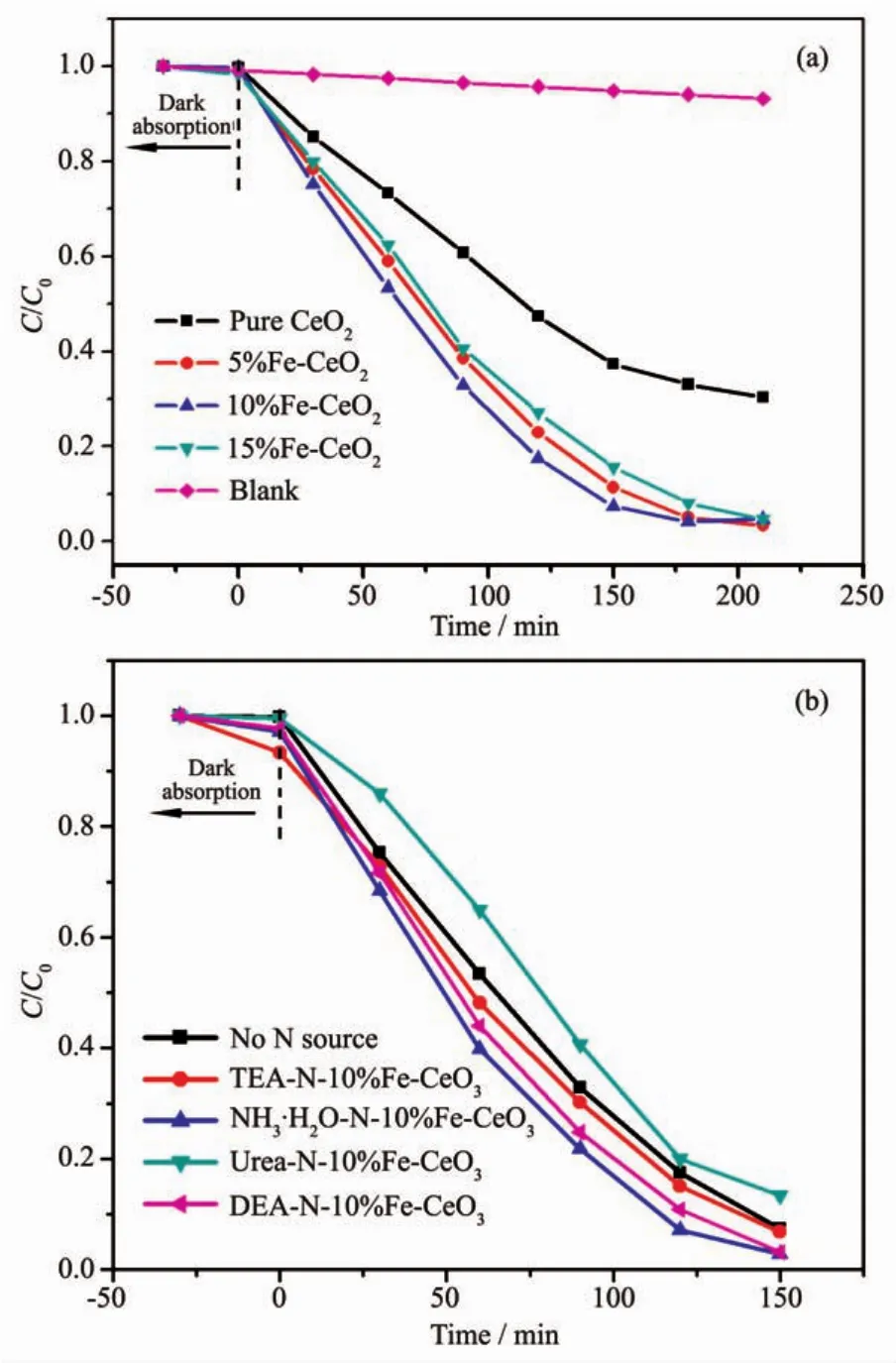
圖6 CeO2和不同含量Fe摻雜的Fe-CeO2(a)及不同氮源N-10%Fe-CeO2(b)作催化劑的光催化降解亞甲基藍溶液性能Fig.6 Photocatalytic degradation of methylene blue solution with pure CeO2and Fe-CeO2as catalystic agents(a)and different nitrogensource N-10%Fe-CeO2as catalytic agents(b)
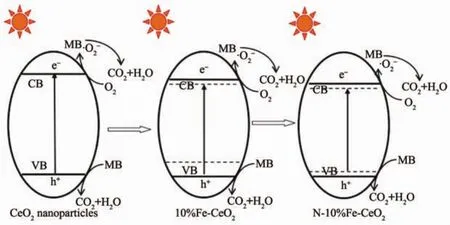
圖 7 CeO2、10%Fe-CeO2和 N-10%Fe-CeO2的催化機理示意圖Fig.7 Schematic diagram of catalytic mechanism of CeO2,10%Fe-CeO2and N-10%Fe-CeO2
2.7 光催化劑的循環使用性能
圖8 為NH3·H2O-N-10%Fe-CeO2的循環使用的降解率結果(每次光催化時間為210 min)。降解率隨著使用次數的增加,略有降低。經過5次使用后,光催化劑在210 min內對亞甲基藍的降解率仍有89%,表明催化劑的光催化性能具有良好的穩定性。圖8(b)對比了5次循環使用后的催化劑與制備態的催化劑的XRD圖,并沒有發現有明顯變化。說明催化劑在使用前后結構沒有改變,也幾乎沒有吸附其它物質。 因此,NH3·H2O-N-10%Fe-CeO2的光催化性能總體上比較穩定。在循環使用過程中,降解率有所下降的原因可能是催化劑在回收再利用過程中發生了微量的質量損失。
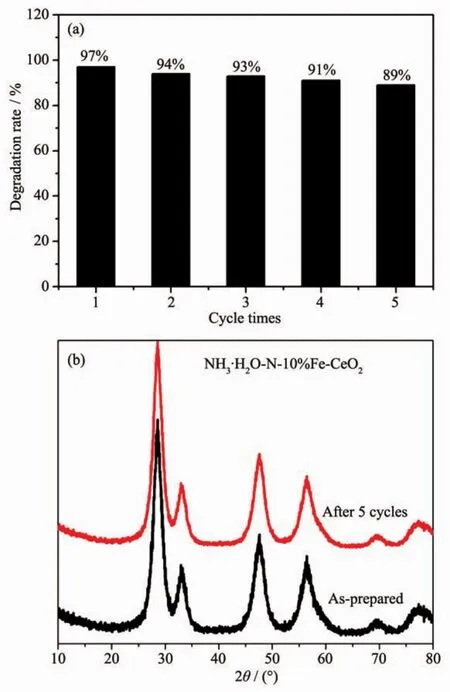
圖8 (a)NH3·H2O-N-10%Fe-CeO2催化劑對亞甲基藍溶液的降解率與循環使用次數的關系;(b)循環使用前后的XRD對比圖Fig.8 (a)Relationship of photocatalytic degradation rate of methylene blue solution and use time with NH3·H2O-N-10%Fe-CeO2as catalytic agent;(b)XRD patterns comparison for as-prepared and after 5 cycles photocatalysis
3 結 論
本文采用溶劑熱法合成了不同Fe摻雜含量的Fe-CeO2納米粉體及不同氮源摻雜的N-10%Fe-CeO2納米粉體,并對其光催化性能進行了研究。可得到以下結論:
(1)Fe摻雜引起晶格畸變,形成反應活性中心,產生雜質能級,降低禁帶寬度。10%Fe-CeO2的催化速率最高,將純CeO2對亞甲基藍的降解率從67%提高到95%。
(2)摻雜過量Fe時,Fe會成為光生電子與空穴的復合中心,從而降低光生載荷的量子效率,降低其催化活性。
(3)不同氮源摻雜對10%Fe-CeO2的生長形貌具有重要影響,其中以濃氨水為氮源摻雜形成中空薄壁狀N-10%Fe-CeO2粉體的禁帶寬度最小,對亞甲藍溶液的降解效率可提高到97%。
(4)以濃氨水為氮源制備的N-10%Fe-CeO2催化劑具有較好的性能穩定性。經5次循環使用,對亞甲基藍溶液的光催化降解率仍高達89%。
參考文獻:
[1]SU Lin-Feng(蘇琳峰),GONG Jin-Feng(鞏金峰),MENG Fan-Ming(孟 凡 明).Chinese Journal of Synthetic Chemistry(合成化學研究),2016,4(4):29-37
[2]QI En-Lei(齊恩磊),MAN Li-Ying(滿麗瑩),WANG Sun-Hao(王孫昊),et al.Chinese Journal of Materials Research(材料研究學報),2011,25(2):219-224
[3]ZHAO Xiao-Bing(趙曉兵),YOU Jing(游靜),LU Xiao-Wang(陸曉旺),et al.J.Inorg.Mater.(無機材料學報),2011,26(2):159-164
[4]WU Jun-Ming(吳俊明),WANG Ya-Ping(王亞平),YANG Han-Pei(楊漢培),et al.Chinese J.Inorg.Chem.(無機化學學報),2010,26(2):203-210
[5]Liyange A D,Perera S D,Tan K,et al.ACS Catal.,2014,4(2):577-584
[6]WANG Jing-Xin(王敬欣).J.Chin.Rare Earth Soc.(中國稀土學報),2007,25(S1):82-88
[7]Wang W,Zhu Q,Dai Q G,et al.Chem.Eng.J.,2017,307:1037-1046
[8]YUAN Qiang(袁強),JIANG Ling(江玲),LI Hui(李輝),et al.Journal of Xiamen University:Natural Sciences Edition(廈門大學學報:自然科學版),2011,50(1):70-75
[9]Arul N S,Mangalaraj D,Chen P,et al.Mater.Lett.,2011,65:3320-3322
[10]Arul N S,Mangalaraj D,Han J.Mater.Lett.,2015,145:189-192
[11]Xie S L,Wang Z L,Cheng F L,et al.Nano Energy,2017,34:313-337
[12]ZHANG Guo-Fang(張國芳),ZHANG Yang-Huan(張羊換),GE Qi-Lu(葛啟錄),et al.Spectrosc.Spectr.Anal.(光譜學與光譜分析),2011,31(12):3315-3318
[13]LI Chang-Quan(李長全),LUO Lai-Tao(羅來濤),XIONG Guang-Wei(熊光偉).Acta Chim.Sin.(化學學報),2010,68(10):1023-1026
[14]GU Ming-Jie(顧明杰),LI Rui-Xing(李銳星),SONG Xiao-Zhen(宋曉貞).Journal of Ceramics(陶瓷學報),2013,34(2):135-138
[15]Mao C J,Zhao Y X,Qiu X F,et al.Phys.Chem.Chem.Phys.,2009,40(3):5633-5638
[16]Wu C L.Mater.Lett.,2015,139:382-384
[17]Ren R K,Zhang M J,Meng J,et al.Chin.Phys.B,2017,26(3):036102
[18]HUANG Dong-Sheng(黃東生),CHEN Chao-Feng(陳朝鳳),LI Yu-Hua(李玉花),et al.Chinese J.Inorg.Chem.(無機化學學報),2007,23(4):738-742
[19]JIAN Li-Xin(翦立新),YIN Xiao-Qiu(殷小秋),XIANG Jian-Nan(向建南),et al.Journal of Hunan University:Natural Sciences Edition(湖南大學學報:自然科學版),2006,33(1):79-82
[20]Sun C W,Li H,Chen L Q.Energy Environ.Sci.,2012,5:8475-8505
[21]Toru Y,Peter H.Appl.Surf.Sci.,2008,254:2441-2449
[22]Jeyanthi C E,Siddheswaran R,Kumar P,et al.Ceram.Int.,2014,40(6):8599-8605
[23]Xu B,Zhang Q T,Yuan S S,et al.Catal.Today,2017,281(1):135-143
[24]Tian D,Zeng C H,Fu Y C,et al.Solid State Commun.,2016,231/232:68-79
