濁點萃取氣相色譜質譜法測定蘋果汁中的有機磷農藥殘留
2017-12-18 11:10:07,
食品工業科技 2017年23期
,
(煙臺工程職業技術學院,山東煙臺 264006)

周璐,成艷娜
(煙臺工程職業技術學院,山東煙臺 264006)
本文對蘋果汁中45種有機磷類農藥的提取、凈化技術和氣相色譜質譜檢測條件進行了研究。以蘋果汁為基質材料,45種有機磷類農藥在1、5、10 μg/L三個水平上對蘋果汁進行添加回收率實驗。結果表明:45種有機磷類目標物在優化條件下的回收率為71.16%~117.94%,相對標準偏差1.20%~15.13%(n=5),方法檢出限為0.01~0.22 μg/L。45種有機磷類農藥能夠實現很好的分離,線性范圍在10~2000 μg/L,線性相關系數大于0.984。本方法操作簡單,結果準確,由于幾乎不使用有機溶劑也減少了分析過程對環境和人體的危害。
濁點萃取,反萃取,氣相色譜質譜法,有機磷類農藥
我國是農藥產量和使用大國,有機磷類占了農藥總數量的大半。有機磷殘留不僅可以通過環境和食物鏈的作用間接對人體健康造成潛在危害,同時也造成一定的環境污染。國內外對于有機磷類農藥殘留的分析報道中,檢測方法主要為氣相色譜法、氣相色譜-質譜聯用法、液相色譜-質譜聯用法、免疫法、酶抑制法等[1-5]。其中,用于檢測前的樣品前處理技術多為固相萃取柱凈化、凝膠滲透色譜凈化、液液萃取、加速溶劑萃取及分散固相萃取等[6-10]。這些方法大都操作時間長,有機溶劑用量大,對環境和人體有一定危害。
濁點萃取技術是一種近幾年新興的環保樣品前處理技術,以表面活性劑水溶液為萃取劑,避免使用有機溶劑提取,通過表面活性劑的增溶性和分相來實現對目標物的分離和富集,最后用微量的有機溶劑將目標物從表面活性劑相中反萃取出來,從而更大的提高了此方法的富集倍數,還可避免表面活性劑粘度較大對檢測儀器的影響。此項技術已廣泛應用于金屬離子、生物大分子、臨床治療藥物檢測、體內微量元素、有毒污染物及中藥成分等樣品的分離分析預處理[11-12],但是關于濁點萃取反萃取法同時富集分離果汁中幾十種有機磷類農藥的研究極少。本文采用的方法具有操作簡便快捷、經濟環保、無需特殊設備等優點。前處理過程幾乎不使用有機溶劑,適用于大批量分析,為保證食品安全和減少分析方法對環境和人體的危害提供了技術支撐。
1 材料與方法
1.1 材料與儀器
蘋果汁 為市售匯源100%純蘋果汁。敵敵畏、乙酰甲胺磷、丙線磷、硫線磷、甲拌磷、甲基乙拌磷、樂果、烯蟲磷、特丁硫磷、殺螟腈、地蟲硫磷、二嗪磷、乙拌磷、氯唑磷、乙嘧硫磷、安果、磷銨、除線磷、甲基毒死蜱、甲基對硫磷、甲基立枯磷、殺螟硫磷、馬拉硫磷、倍硫磷、毒死蜱、對硫磷、水胺硫磷、甲基溴硫磷、異柳磷、毒蟲畏、喹硫磷、稻豐散、丙蟲磷、乙基溴硫磷、抑草磷、丙硫磷、丙溴磷、乙硫磷、三唑磷、硫丙磷、三硫磷、苯腈磷、敵瘟磷、苯硫磷、付殺硫磷 均購于德國Dr. Ehrenstorfer公司。PEG 2000、PEG 4000、PEG 6000、PEG 8000、正丁醇、無水硫酸鈉、正己烷、異辛烷、石油醚 天津博迪化工股份有限公司試劑。
QL-886漩渦混合器 海門市其林貝爾儀器有限公司;Agilent 7890A/5975C氣相色譜質譜儀 美國安捷倫公司;AS3120超聲儀 中西遠大科技有限公司;FA2004電子天平(0.0001 g) 上海天平儀器廠;彩色單道移液槍 美國瑞寧儀器公司。
1.2 實驗方法
1.2.1 農藥標準溶液的配制 分別取適量的45種有機磷類農藥標準品,用正己烷-丙酮(7∶3,v/v)定容配制成質量濃度100 mg/L的標準溶液,再用正己烷-丙酮(7∶3,v/v)逐級稀釋成2、0.5、0.1、0.02、0.01 mg/L的混合標準儲備液,于4 ℃下保存。
1.2.2 樣品前處理技術 準確移取10.0 mL蘋果汁于15 mL刻度試管中,依次準確加入3.0 mL 300 g/L PEG 4000溶液、1.10 g無水硫酸鈉,混合均勻并超聲輔助全部溶解后,準確加入0.4 mL正丁醇,混合均勻。將上述混合溶液于60 ℃條件下水浴20 min。待其降至室溫后移取下層水相棄去。向刻度試管中剩下的表面活性劑相中準確加入250 μL正己烷,漩渦混合1 min,待其分層后吸取上層正己烷相移至自動進樣小瓶,待測。
1.2.3 GC-MS分析條件 4 Agilent 7890A/5975C氣相色譜儀,VG Trio-2000質譜檢測器;色譜柱:HP-5 MS柱(30 m×0.25 mm×0.25 μm);升溫程序為:70 ℃(保持2 min)以25 ℃/min的速度升至145 ℃,再以3 ℃/min的速度升至200 ℃,以8 ℃/min的速度升至280 ℃(保持5 min);在SIM模式下檢測,采用電子轟擊源(EI+);電壓為70 eV;離子源溫度為230 ℃;流速:1 mL/min;載氣:氦氣(純度99.999%);進樣量:1 μL。
1.2.4 方法檢出限的測定 在空白樣品中添加標準溶液進行檢測,以三倍信噪比(S/N=3)對應的濃度為方法檢出限。
2 結果與分析
2.1 濁點萃取條件優化
濁點萃取的操作過程是在樣品液中加入表面活性劑溶液、無機鹽、醇類及其他成分,混合均勻,然后將混合液在適當的溫度下平衡一段時間,待混合溶液中兩相完全分離后,棄去水相,富集后的表面活性劑相適當處理后待分析[13-14]。
前處理過程中,表面活性劑的種類及其用量、添加劑的用量、平衡溫度及平衡時間是影響萃取效果的幾個最主要因素。其中表面活性劑的種類及用量直接決定了目標藥物的有效提取率,關系著檢測結果的準確性。同時這幾個參數又互相影響,只有全部優化了這幾個參數才能得到理想的富集、凈化效果和回收率。
2.1.1 表面活性劑種類的選擇 濁點萃取效率直接受表面活性劑疏水和親水兩部分的影響。同時各種表面活性劑都有一定的適用對象,本實驗選擇4種HLB值不同且能與反萃取溶劑正己烷分層的表面活性劑(PEG 2000、PEG 4000、PEG 6000、PEG 8000)作為優化對象進行研究。PEG 2000需要的水浴溫度過高(超過60 ℃),因而重點對PEG 4000、PEG 6000和PEG 8000進行比較,結果PEG 4000的回收率范圍是69.6%~119.4%,PEG 6000的回收率范圍是65.9%~121.7%,PEG 8000的回收率范圍是64.8%~124.1%。由圖1可以看出,PEG 4000作為濁點萃取的表面活性劑時添加回收率接近100%的農藥種類最多。

圖1 表面活性劑種類對目標物回收率的影響Fig.1 Effect of types of surfactant on recovery
2.1.2 表面活性劑用量的選擇 表面活性劑的用量顯著影響回收率、濃縮因子、相比等參數。用量少會出現回收率低、準確度和重現性欠佳等問題,嚴重影響實驗定量研究。用量過多,也可能出現回收率降低的現象,同時會在進樣過程中產生負面影響,如目標物保留時間發生變化等[15]。
因此本實驗在PEG 4000的濃度為300 g/L時進行研究。PEG 4000濃度太高時溶液粘稠不利于吸取轉移;濃度太低需要的體積過大時會降低富集倍數。當PEG 4000添加量為2.5 mL時回收率范圍是65.9%~116.2%,添加量為3.0 mL時回收率范圍是73.4%~118.3%,添加量為3.5 mL時回收率范圍是69.1%~123.5%。結果如圖2所示,當PEG 4000添加量為3.0 mL時45種有機磷的回收率接近100%的數量最多,能夠滿足農藥殘留分析的要求。

圖2 PEG 4000用量對目標物回收率的影響Fig.2 Effect of concentration of PEG 4000 on recovery
2.1.3 正丁醇量的優化 萃取過程中加入適當的添加劑可以改變表面活性劑的濁點溫度,引發表面活性劑的相分離。本研究中,正丁醇的添加量對45種有機磷類農藥的回收率有一定的影響。在300~450 μL范圍內對正丁醇的用量進行優化。當正丁醇添加量為300 μL時回收率范圍是65.6%~111.4%,添加量為350 μL時回收率范圍是66.9%~117.2%,添加量為400 μL時回收率范圍是74.9%~113.6%,添加量為450 μL時回收率范圍是72.5%~119.7%。結果表明:正丁醇的添加量為400 μL時,回收率接近100%的農藥種類最多。

圖3 正丁醇用量對目標物回收率的影響Fig.3 Effect of the volume n-butyl alcohol on recovery
2.1.4 鹽種類及用量的優化 添加惰性鹽于溶液中可以增大溶液離子強度,使水相密度增大,加速兩相分離;降低表面活性劑的濁點溫度,還可以增加目標物的回收率。
本實驗經初步預選后使用無水Na2SO4和無水MgSO4進行添加回收率實驗。由圖4可以看出,在添加量相同的條件下,添加無水Na2SO4比添加無水MgSO4時農藥的平均回收率接近100%的數量多且較穩定,選擇無水Na2SO4做進一步用量優化實驗。考察了0.9~1.2 g無水硫酸鈉的添加量對濁點萃取的影響。結果表明:1.10 g無水硫酸鈉的添加量回收率最理想(圖5)。

圖4 鹽的種類對目標物回收率的影響Fig.4 Effect of the salt type on recovery

圖5 無水硫酸鈉的添加量對目標物回收率的影響Fig.5 Effect of the mass of sodium sulfate anhydrous on recovery
2.1.5 平衡溫度的優化 采用非離子表面活性劑萃取目標物時,提高平衡溫度會導致非離子表面活性劑與水作用的氫鍵破壞而發生脫水現象,從而使萃取率增加,富集相體積減小[13]。通常,采用的平衡溫度高于濁點以上15~20 ℃。由此本實驗在50~70 ℃范圍內對平衡溫度進行優化,圖6為當表面活性劑為PEG 4000,其濃度為300 g/L,正丁醇添加量為0.4 mL,無水硫酸鈉添加量為1.1 g時,平衡溫度對目標物回收率的影響情況。實驗結果顯示:在60 ℃時,45種有機磷類農藥的回收率接近100%的種類最多。

圖6 平衡溫度對目標物回收率的影響Fig.6 Effect of equilibration temperature on recovery
2.1.6 平衡時間的優化 濁點萃取中存在表面活性劑、單體和待測物在水相、膠束相兩個平衡過程,這兩個過程都需要一定的時間達到平衡。前者平衡時間與環境溫度又有密切關系[14]。由此本實驗在10~40 min范圍內對平衡時間進行優化。圖7為當表面活性劑為PEG 4000,其濃度為300 g/L,正丁醇添加量為0.4 mL,無水硫酸鈉添加量為1.1 g時,平衡溫度為60 ℃時,平衡時間對目標物回收率的影響。實驗結果表明:平衡20 min,45種有機磷類農藥的回收率接近100%的種類最多。因此,選擇20 min作為最優平衡溫度來進一步完成實驗研究。

圖7 平衡時間對目標物回收率的影響Fig.7 Effect of time on recovery
2.2 反萃取條件優化
2.2.1 反萃取溶劑的選擇 為增加萃取富集率,降低檢出限數值。本實驗考慮采用既能與水相分層又對有機磷有較高溶解度的二氯甲烷、乙酸乙酯、正己烷、異辛烷、石油醚作為反萃取溶劑。由于二氯甲烷萃取后在下層不利于吸取進樣,乙酸乙酯與表面活性劑相不分層,故先排除這兩種溶劑。用正己烷、異辛烷、石油醚做添加回收率實驗,結果表明:正己烷的平均回收率接近100%的農藥種類最多且符合農殘分析要求(圖8)。
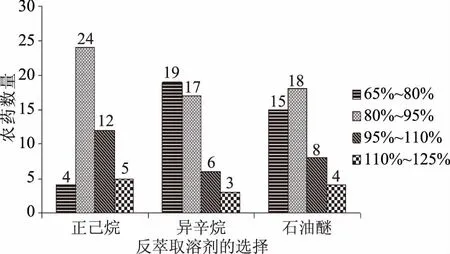
圖8 反萃溶劑的選擇Fig.8 The type of reverse extraction solvent

表1 標準溶液的線性范圍、線性方程、相關系數和檢出限Table 1 Linear range,linear equations,correlation coefficients and LOD for the standard mixture of pesticides
2.2.2 反萃取溶劑用量的選擇 考察正己烷體積對回收率的影響,當正己烷體積低于200 μL時,雖然富集倍數較高但由于體積小會給操作帶來不便,容易吸取到表面活性劑層進樣后會對儀器造成損傷;當體積過大時被測組分的富集倍數會越小[15]。故在200~350 μL范圍內考察正己烷用量對回收率的影響,根據回收率接近100%的農藥種類數量最終確定正己烷的用量為250 μL(圖9)。

圖9 正己烷用量的選擇Fig.9 The concentration of n-hexane
2.3 方法的靈敏度和精密度
將45種有機磷農藥配制成2000、500、100、20、10 μg/L的混合標準溶液按上述優化好的CPE-GC/MS法,檢測得到線性范圍、線性方程、相關系數、方法檢出限,如表1。

續表

表2 45種有機磷類農藥的添加回收率及相對標準偏差(n=5)Table 2 Spiked,recovery and precision(n=5)

續表
在1、5、10 μg/L三個水平上進行添加回收率實驗,每一添加水平重復5次實驗,外標法定量。利用空白樣品標定校正計算的回收率和相對標準偏差見表2,表明該方法有良好的精密度和準確度,能夠達到痕量分析的要求。在優化的色譜及質譜條件下45種有機磷類農藥能夠得到很好的分離,峰形良好。45種農藥標準物質的總離子流圖見圖10。當樣品的添加水平為5 μg/L時,45種有機磷類農藥的總離子流色譜圖見圖11。
圖10 45種有機磷類農藥標準物質的總離子流色譜圖 Fig.10 GC-MS total ion current chromatograms of the mixed standard solution of 45 organophosphorous pesticides注:標準溶液濃度為0.5 mg/L。

圖11 樣品中45種有機磷類農藥的總離子流色譜圖Fig.11 GC-MS total ion current chromatograms of 45 organophosphorous pesticides 注:添加水平為5 μg/L。
3 結論
本實驗選擇蘋果汁為供試樣品,45種廣泛使用的有機磷類農藥(敵敵畏、甲拌磷、樂果等)為待測農藥,通過前處理條件的優化建立了濁點萃取-正己烷反萃取氣相色譜質譜聯用法分析檢測蘋果汁中有機磷類農藥的技術。確定了以300 g/L PEG 4000水溶液為萃取劑,無水硫酸鈉、正丁醇的添加量為1.10 g和400 μL,平衡溫度和時間為60 ℃和20 min的濁點萃取因素。反萃取溶劑選擇正己烷,用量為250 μL。利用此技術在1、5、10 μg/L三個水平上對供試蘋果汁進行添加回收率實驗,實驗結果表明:45種有機磷類目標物在優化條件下的回收率為71.16%~117.94%,相對標準偏差1.20%~15.13%,實驗操作簡單,結果準確,精密度高。在10~2000 μg/L范圍內,45種有機磷類農藥線性關系良好,相關系數均大于0.984;LOD值為0.01~0.22 μg/L,均低于國家規定最大殘留限量標準和日本肯定列表中農作物種農藥殘留的一律標準(10 μg/L)的限量要求[16],同時45種有機磷類農藥得到了很好的分離,峰型對稱,避開了蘋果汁中雜質的干擾。本方法簡化了樣品的前處理的步驟,節約了分析時間,靈敏、高效,適用于大批量分析,且極少使用有機溶劑,經濟環保。采用反萃取操作后將農藥的富集率從3.3倍提升到40倍,降低了檢出限。本方法為保證食品安全,減少對環境和人體的危害提供了技術保障,且適用廣泛。
[1]湯婕,湯鋒,岳永德,等.氣相色譜法(GC-FPD)快速測定蔬菜、水果中18種有機磷農藥殘留[J].安徽農業大學學報,2013,40(6):1054-1058.
[2]周蓉,曹趙云,趙肖華,等.分散固相萃取-分散液液微萃取/氣相色譜-串聯質譜法測定蔬菜中19 種有機磷農藥殘留[J].分析測試學報,2017,36(1):67-72.
[3]劉琪,孫雷,張驪. 超高效液相色譜-串聯質譜法測定豬肝中有機磷農藥殘留量的研究[J].分析測試學報,2010,29(7):747-750.
[4]Domotorova Milena,Matisova Eva,Kirchner Michal,et al. MSPD combined with fast GC for ultratrace analysis of pesticide residues in non-fatty food[J]. Acta Chimica Slovenica,2005,52(4):422-428.
[5]Xu Dun-ming,Yang Fang,Lu Sheng-yu,et al. Determination of indoxacarb residue in foodstuffs of plant and animal origin by GC-ECD and LC-MS/MS[J]. Agricultural Sciences in China,2008,7(10):1228-1234.
[6]張仕云,鄭仲華,丘峰,等.氣相色譜-質譜法快速測定葡萄酒中7種有機磷農藥[J].食品與發酵科技,2016,52(6):91-95.
[7]何超,劉躍華,黃海濤,等.氣相色譜法測定煙草中有機磷農藥殘留量[J]. 理化檢驗(化學分冊),2010,46(6):663-668.
[8]潘守奇,孫軍,董靜,等.氣相色譜法同時測定蔬菜中24種有機磷農藥殘留[J].分析實驗室,2010,29(1):115-118.
[9]Anastassiades Michelangelo,Lehotay Steven J,Stajnbaher Darinka,et al. Fast and easy multiresidue method employing acetonitr ile extraction/partitioning and dispersive solid-phase extraction for the determination of pesticide residues in product[J]. J AOAC International,2003,86(2):412-431
[10]Caro E,Marce R M,Cormack P A G,et al. Novel Enrofloxacin imprinted polymer applied to the solid phase extraction. Fluorinated quinolones from urine and tissue samples[J]. Analytical Chimica Acta,2006,562(2):145-151.
[11]Aline oriano opes,Jerusa Simone Garcia,Rodrigo Ramos Catharino. Cloud point extraction applied to casein proteins of cow milk and their identification by masss pectrometry[J]. Analytica Chimica Acta,2007,590:166-172.
[12]Alireza Rezaie Rod,Shahin Borhani,Farzaneh Shemirani. Cloud point preconcentration and flame atomic absorption spectrometry:application to the determination of manganese in milk and water samples[J]. Eur Food Res Technol,2006,223:649-653.
[13]肖珊美,羅小會.濁點萃取-酶抑制-分光光度法測定蔬菜中有機磷農藥殘留量[J].理化檢驗化學分冊,2015,51(5):713-715.
[14]李文廷,向楊華,王靜.濁點萃取-氣相色譜質譜法檢測中藥材中的有機磷農藥殘留[J].云南化工,2016,43(4):64-67.
[15]莫小榮,鄭春慧,陳建偉,等.濁點萃取-異辛烷反萃取-氣相色譜測定茶葉中擬除蟲菊酯農藥殘留[J].分析化學研究報告,2009,37(8):1178-1182.
[16]周宏琛,朱濤,王勇,等.“日本肯定列表制度”農藥殘留新標準對我國農產品出口的影響[J]. 現代食品科技,2006,22(4):197-199.
DeterminationoforganophosphorouspesticidesinciderbyGC-MScoupledwithcloudpointextraction
ZHOULu,CHENGYan-na
(Yantai Engineering and Technology College,Yantai 264006,China)
This article focused on the technique using GC-MS to determine the presence and concentrations of 45 kinds of organophosphorous pesticides in cider. 45 pesticides were tested with the recovery experiment of cider at three levels:1,5,10 μg/L.The results showed that the 45 pesticides gave satisfactory recoveries ranging from 71.16% to 117.94% with the RSDs from 1.20% to 15.13%(n=5),the limitation of this method was between 0.01 μg/L and 0.22 μg/L. 45 pesticides were well separated and the linear relationship was good enough which the correlation coefficients were greater than 0.984 for concentrations from 10 to 2000 μg/L for most pesticides. Compared with conventional techniques,it was more accurate,low-cost,highly efficient and environmental friendly. Furthermore,for the limited use of organic solution,it has little harm to environment and human’s health for the limited use of organic solution during the analysis process.
cloud point extraction;back-extraction;gas chromatography-mass spectrometer;organophosphorus pesticides
2017-06-12
周璐(1986-),女,碩士,研究方向:食品營養與檢測,E-mail:297371859@qq.com。
TS207.3
A
1002-0306(2017)23-0225-07
10.13386/j.issn1002-0306.2017.23.042
