間充質干細胞緩解大鼠急性肺損傷的分子機制
2017-09-03 02:32:24胡曉旻王延安
中國老年學雜志 2017年15期
關鍵詞:水平
胡曉旻 劉 剛 劉 超 王延安
(天津市第三中心醫院心臟中心,天津 300170)
間充質干細胞緩解大鼠急性肺損傷的分子機制
胡曉旻 劉 剛1劉 超2王延安3
(天津市第三中心醫院心臟中心,天津 300170)
目的 探討間充質干細胞(MSCs)抑制核因子(NF)- κB的活性和抑制親環素(CyP)A的表達而緩解急性肺損傷(ALI)的分子機制。方法 首先比較臍帶和骨髓MSCs的細胞形態、細胞表型、分化能力和免疫能力,并采用全基因組表達芯片比較兩者的功能基因表達差異。通過注射脂多糖(LPS)制備SD大鼠ALI模型,研究輸注MSCs能否通過抑制CyPA的表達和抑制NF- κB的活性,進而控制炎癥反應的過度激活和抑制氧化應激等,減輕內毒素所致的ALI。 結果 CD29在臍帶MSCs的陽性表達率低于在骨髓MSCs中的表達率(P<0.01)。聚類分析發現骨髓來源的MSCs中高表達的基因主要集中在免疫相關和骨骼發育相關的基因,而臍帶MSCs中高表達的基因主要集中在細胞分化、器官發育和信號轉導的相關基因。對SD大鼠ALI損傷模型的研究發現,MSCs干預可減輕LPS對肺組織的損傷程度。LPS組各時間點血漿促炎因子巨噬細胞炎癥蛋白(MIP)- 2 水平顯著高于對照組和MSCs+LPS組(P<0.05)。MSCs可顯著抑制LPS誘發的炎癥因子MIP- 2的增高(P<0.05)。與對照組相比,LPS組在各時間點CyPA蛋白表達和TNF- α表達水平均顯著升高(P<0.05),而MSCs可以抑制CyPA的蛋白和TNF- α的表達(P<0.05)。肺組織NF- κB 在LPS組增高,MSC干預可有效降低NF- κB 表達(P均<0.05)。LPS組肺組織丙二醛(MDA)和髓過氧化物酶(MPO)活性水平在三個時間點均明顯高于對照組,在各相應時間點,MSCs+LPS組肺組織MDA和MPO均明顯低于LPS組(P均<0.05)。結論 臍帶MSCs明顯降低了大鼠血漿促炎因子的水平,可顯著抑制CyPA的表達,并通過抑制NF- κB的活性減輕了LPS引起的全身炎癥反應和氧化應激,減輕了LPS所致的肺損傷。
間充質干細胞;急性肺損傷;核因子- κB;親環素A;信號轉導通路
急性肺損傷(ALI)發病機制目前仍未闡明,目前臨床上ALI的治療主要是支持性治療,尚未找到針對性的治療措施。存在于革蘭陰性菌表面的脂多糖(LPS)可以通過Toll樣受體(TLR)4激活下游的核因子(NF)- κB信號通路〔1,2〕。NF- κB信號通路活化后,啟動靶基因的表達,可以引起腫瘤壞死因子(TNF)- α、白細胞介素(IL)- 1、IL- 6、IL- 8等多種炎性介質及細胞因子的釋放,誘發炎癥反應。親環素(CyP)A在細胞內廣泛存在,生理功能為對單核細胞、巨噬細胞、內皮細胞和平滑肌細胞產生氧化刺激,而且在細胞外部對白細胞具有趨化作用。CyPA通過與受體CD147結合,可以活化NF- κB通路〔3〕,因此在病理情況下具有促炎作用。間充質干細胞(MSCs)具有多向分化、免疫調節的特性,不僅能夠歸巢至損傷組織,抑制受損組織發生的免疫反應和過度炎性反應,MSCs還具有向多種組織、細胞分化的潛能,因此在創傷修復、組織功能再生與重建、免疫調節等領域應用前景廣闊〔4〕。MSCs可通過其抗炎、抗凋亡和免疫調節的作用,減輕內毒素引起的ALI,改善肺功能,提高ALI/急性呼吸窘迫綜合征(ARDS)模式動物的生存率,為ALI的治療提供了新的方法與思路。本研究探討抑制NF- κB的活性和CyPA的表達而緩解ALI的分子機制。
1 資料和方法
1.1 材料與試劑 臍帶MSCs的獲取:采集足月健康新生兒臍帶組織30份。足月正常分娩之臍帶來自我院產科病房,均取得產婦及家屬的知情同意。產婦的年齡為(29.1±5.7)歲。骨髓MSCs的獲取:常規局麻,于髂骨取骨髓,分離制備骨髓MSCs。低糖DMEM培養基、高糖DMEM培養基購自武漢博士德生物工程有限公司;胎牛血清和Trizol試劑購自Gibco公司。熒光標記鼠抗人CD29、CD44、CD73、CD105和CD166及其同型對照購自Sigma公司。
1.2 MSCs的培養和鑒定 臍帶MSCs采用胰酶消化法獲取。取無菌臍帶2 cm,磷酸鹽緩沖液(PBS)洗凈,剪碎后使用Ⅳ型膠原酶37℃消化,離心,洗滌3遍,取下層沉淀,再以胰蛋白酶PBS消化后離心,獲取沉淀后使用吹打細胞懸浮,以100 μm濾網過濾并離心獲得單細胞。然后以PBS離心后洗滌細胞2次,置于低糖DMEM培養基中培養。骨髓組織用PBS液稀釋后,放置在淋巴細胞分離液面上,離心25 min后獲得單個核細胞,采用DMEM培養基培養。另種MSCs均傳至F4代時進行常規倒置顯微鏡觀察,進行HE染色。在含MSCs的PBS中加入鼠抗人抗體PE- CD44,FIFC- CD29,PE- CD73,FIFC- CD90,PE- CD105,PE- CD166,進行流式細胞檢測。
1.3 利用基因芯片分析MSCs的基因表達 使用人全基因組表達基因芯片對這兩種細胞的基因組表達和微小表達情況進行比較分析,并對差異表達的基因進行功能聚類分析。采用Affymetrix mrcroarray 公司提供的基因差異表達芯片,生物素標記cDNA,進行雜交反應,采用基因芯片操作軟件對數據進行采集和處理,使用DAVID分析軟件進行功能聚類分析。
1.4 膿毒癥ALI模型的制作及輸注MSCs懸液治療肺損傷 選擇SD大鼠90只,隨機分成3組,分別為對照組30只、LPS組30只和MSC+LPS組30只。LPS組和MSC+LPS組均采用腹腔內注射LPS 10 mg/kg的方法制作膿毒癥模型,后者于LPS注射1 h后,通過鼠尾靜脈給予含5×105臍帶MSCs的生理鹽水300 μl,對照組和LPS組則給予300 μl生理鹽水。分別于尾靜脈注射后6 h、24 h和48 h處死大鼠,每次10只,于左肺取得支氣管肺泡灌洗液,測定支氣管肺泡灌洗液蛋白濃度,收集血漿和右肺組織。取右肺下葉,采用4%多聚甲醛溶液對SD大鼠肺臟進行固定,常規病理切片后進行HE染色。以肺泡充血、出血、中性粒細胞浸潤和肺泡間隔增厚或透明膜形成等四種病變對肺組織損傷進行綜合評定并分級。0級為無損傷,1級肺組織損傷面積小于1/4,2級肺組織損傷面積介于1/4~1/2,3級肺組織損傷面積介于1/2~3/4,4級肺組織損傷面積大于3/4。取右肺中葉,制備肺組織勻漿。將各組肺組織勻漿100 mg,ELISA方法測定肺組織TNF- α水平,免疫熒光法測定肺組織CyPA蛋白含量,并測定CyPA mRNA表達水平。取右肺上葉,先稱重,然后進行烘干并再次稱重,計算肺組織濕干比。采用ELISA的方法測定炎癥因子巨噬細胞炎癥蛋白(MIP)- 2、IL- 10、CyPA和NF- κB水平。采用Western印跡方法測定NF- κB的肺組織入核水平。對肺組織丙二醛(MDA)和髓過氧化物酶(MPO)活性進行測定。
1.5 統計學方法 采用SPSS19.0統計軟件進行方差分析。
2 結 果
2.1 兩種MSCs細胞表面抗原表達 臍帶來源的MSCs細胞表面CD44、CD73、CD90、CD105和CD166陽性表達率分別為(92.15±4.31)%、(94.43±3.68)%、(97.39±4.74)%、(93.88±5.11)%和(98.62±4.64)%,骨髓來源的MSCs細胞表面CD44、CD73、CD90、CD105和CD166陽性表達率分別為(92.78±4.22)%、(95.02±3.49)%、(96.86±4.45)%、(93.23±4.98)%和(98.23±4.14)%,兩組相比差別無統計學意義。但是CD29在臍帶MSCs的陽性表達率僅(43.5±2.32)%,明顯低于在骨髓MSCs中的表達率(90.15±3.43)%(P<0.01)。
2.2 基因芯片分析MSCs的基因表達 基因芯片mRNA差異表達分析臍帶來源MSCs和骨髓來源MSCs之間至少4倍差異表達的基因,可得到1 190個差異表達的基因。聚類分析發現骨髓來源的MSCs中高表達的基因主要集中在免疫相關和骨骼發育相關的基因,而臍帶MSC中高表達的基因主要集中在細胞分化、器官發育和信號轉導的相關基因。
2.3 SD大鼠肺組織病理、肺組織濕干比、支氣管肺泡灌洗液蛋白濃度改變 SD大鼠于腹腔注射LPS 6 h開始,肺組織病理開始肺毛細血管充血、出血,肺毛細血管、肺泡間隔和肺泡腔可見大量炎性細胞浸潤,部分肺泡內可見富含蛋白質的水腫液,于LPS注射24 h時達到高峰,48 h水腫液開始減少。MSC+LPS組肺組織的損傷程度于6 h、24 h和48 h三個時間點均較LPS組明顯減輕。LPS組24 h肺組織濕干比明顯升高并達峰,MSC+LPS組24 h肺干濕比明顯低于LPS組(P<0.05),48 h MSC+LPS組和LPS比較,肺組織濕干比差異無統計學意義(P>0.05)。支氣管肺泡灌洗液蛋白濃度在LPS組明顯增高升高,并于24 h達到高峰。與LPS組比較,MSC+LPS組在各時間點支氣管肺泡灌洗液蛋白濃度差異無統計學意義(P>0.05)。
2.4 不同時間點血漿MIP- 2、IL- 10 水平的組間比較 LPS組在時間點血漿促炎因子MIP- 2 水平顯著高于對照組和MSC+LPS組(P<0.05);血清IL- 10 水平組間比較差異無統計學意義(P>0.05)。MIP- 2水平在LPS刺激下明顯升高并于6 h達峰。MSC+LPS組各時間點MIP- 2水平顯著低于LPS組,說明MSCs可顯著抑制LPS誘發的炎癥反應。見表1。
2.5 肺組織CyPA、TNF- α 水平的組間比較 與對照組相比,LPS組在6 h、12 h和24 h CyPA蛋白表達均顯著升高(P<0.05),與LPS組相比,MSC+LPS組CyPA表達下降有統計學意義(P<0.05)。說明LPS可以明顯提高SD大鼠肺組織的CyPA的表達,而MSCs可以抑制CyPA的蛋白表達。LPS組肺組織TNF- α表達水平在各時間點均高于對照組的表達水平(P<0.05),各相應時間點MSC+LPS組TNF- α表達水平均低于LPS組(P<0.05)。見表2。
2.6 肺組織NF- κB(P65)入核檢測結果的比較 肺組織NF- κB在對照組、LPS組和MSC+LPS組NF- κB 表達分別為(0.25±0.04)、(0.69±0.04)和(0.42±0.04),三組之間比較差異均有統計學意義(P<0.05)。
2.7 肺組織MDA和MPO活性水平比較 LPS組肺組織MDA在三個時間點均明顯高于對照組(P<0.05);在各相應時間點,MSC+LPS組肺組織MDA水平則明顯低于LPS組(P<0.05)。三個時間點LPS組MPO活性水平明顯高于對照組(P<0.05);MSC+LPS組明顯低于LPS組(P<0.05);各組內MPO水平差異無統計學意義(P>0.05),見表3。

表1 不同時間點各組血漿MIP- 2、IL- 10水平比較
與LPS 組比較:1)P<0.05;與對照組比較:2)P<0.05,下表同

表2 不同時間點肺組織CyPA、TNF- α 水平的比較
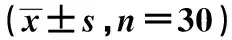
表3 各組不同時間點肺組織MDA和MPO活性水平比較
3 討 論
膿毒癥所致的ALI發病具體機制目前尚未完全明確。一過性的炎癥反應是機體控制感染的自我保護反應,目前普遍認為ALI發病的本質是大量炎癥細胞和炎癥因子導致的過度失控性的炎性反應。中性粒細胞、巨噬細胞在內毒素作用下,產生TNF- α、白細胞介素等促炎因子,啟動炎性反應的級聯反應〔5〕。因此內毒素血癥是ALI/ARDS發病的始動關鍵環節,通過活化各種炎癥細胞,觸發炎癥反應,進而導致肺血管通透性增高、肺泡腔蛋白質滲出、肺水腫和透明膜的形成。
既往研究表明,LPS可以TLR4激活下游的NF- κB信號通路〔6〕。NF- κB作為核轉錄因子,通過與B細胞免疫球蛋白的輕鏈κ鏈基因增強子序列結合,調節靶基因表達,引起包括MIP- 2、TNF- α和IL家族在內的多種炎性介質和細胞因子釋放,從而誘發機體的炎癥反應,因此NF- κB通路在機體炎癥反應過程中起核心作用〔2,6〕。因此阻斷NF- κB信號轉導通路可減輕LPS誘導的ALI的炎癥反應,因此有望成為治療ALI/ARDS的靶點。MSCs可以從骨髓、臍帶、脂肪、肝臟等臟器或組織獲得,然而不同組織取得的MSCs在功能和分化上不盡相同。本研究結果證實臍帶來源的MSCs具有更高的增殖率,骨髓來源的MSCs中高表達的基因主要集中在免疫相關和骨骼發育相關的基因,而臍帶MSCs中高表達的基因主要集中在細胞分化、器官發育和信號轉導的相關基因。對于盲腸結扎穿孔的膿毒癥小鼠模型的研究表明受累的肺組織中MIP- 2 表達及MPO 均明顯增高〔7〕。本研究說明MSCs干預可明顯減輕大鼠肺損傷。
炎癥因子會進一步活化中性粒細胞和血管內皮細胞,趨化中性粒細胞遷移到肺組織內。中性粒細胞激活后產生氧自由基和蛋白酶,中性粒細胞呼吸爆發,可直接損傷肺組織或通過激活誘導炎癥因子釋放,引發炎癥逐級放大〔8〕。MPO可反映中性粒細胞的浸潤程度〔9〕,而MDA是脂質過氧化的產物〔10〕。本研究說明MSCs干預可阻斷或部分阻斷中性粒細胞的浸潤和呼吸爆發。
CyPA 主要由活性氧化物質的激活分泌,通過與受體CD147結合激活NF- κB 通路,調節促炎因子TNF- α等的釋放〔3,11〕。NF- κB以p65/p50二聚體的形式存在于胞質中,其中p65含有轉錄活化區域。靜息狀態下,胞質中的NF- κB與IκB- α結合,使p65的轉錄活化區域功能受到抑制而不能發揮轉錄調節作用〔12〕。本研究說明MSCs在NF- κB通路的上游起作用,從而干預caspase級聯放大的炎癥反應。
1 伍冬冬,潘頻華,覃慶武.急性肺損傷/急性呼吸窘迫綜合征發病機制研究進展〔J〕.中華結核和呼吸雜志,2015;38(7):524- 7.
2 Sender V,Stamme C.Lung cell- specific modulation of LPS- induced TLR4 receptor and adaptor localization〔J〕.Commun Integr Biol,2014;7:e29053.
3 Zhu D,Wang Z,Zhao JJ,etal.The Cyclophilin A- CD147 complex promotes the proliferation and homing of multiple myeloma cells〔J〕.Nat Med,2015;21(6):572- 80.
4 Zhou Z,You Z.Mesenchymal stem cells alleviate LPS- induced acute lung injury in mice by MiR- 142a- 5p- controlled pulmonary endothelial cell autophagy〔J〕.Cell Physiol Biochem,2016;38(1):258- 66.
5 Standiford TJ,Ward PA.Therapeutic targeting of acute lung injury and acute respiratory distress syndrome〔J〕.Transl Res,2016;167(1):183- 91.
6 He X,Wei Z,Zhou E,etal.Baicalein attenuates inflammatory responses by suppressing TLR4 mediated NF- κB and MAPK signaling pathways in LPS- induced mastitis in mice〔J〕.Int Immunopharmacol,2015;28(1):470- 6.
7 Aratani Y,Miura N,Ohno N,etal.Role of neutrophil- derived reactive oxygen species in host defense and inflammation〔J〕.Med Mycol J,2012;53(2):123- 8.
8 Jennette JC,Falk RJ.Pathogenesis of antineutrophil cytoplasmic autoantibody- mediated disease〔J〕.Nat Rev Rheumatol,2014;10(8):463- 73.
9 Flint SM,McKinney EF,Smith KG.Emerging concepts in the pathogenesis of antineutrophil cytoplasmic antibody- associated vasculitis〔J〕.Curr Opin Rheumatol,2015;27(2):197- 203.
10 Ho E,Galougahi K,Liu CC,etal.Biological markers of oxidative stress:applications to cardiovascular research and practice〔J〕.Redox Biol,2013;1(10):483- 91.
11 Shibata N,Kobayashi M.The role for oxidative stress in neurodegenerative diseases〔J〕.Brain Nerve,2008;60(2):157- 70.
12 Dyson HJ,Komives EA.Role of disorder in IκB- NFκB interaction〔J〕.IUBMB Life,2012;64(6):499- 505.
〔2015- 12- 31修回〕
(編輯 曹夢園)
2015年中國博士后科學基金資助項目(2015M581308);天津市科學技術委員會重點項目(11ZCGYSY02000);天津市衛生局重點攻關項目(12KG106)
劉 剛(1972- ),男,醫學碩士,主治醫師,主要從事臨床麻醉、疼痛診療和緩和醫療研究。
胡曉旻(1974- ),男,博士,副主任醫師,主要從事心臟重癥治療與急救研究。
R563
A
1005- 9202(2017)15- 3664- 04;
10.3969/j.issn.1005- 9202.2017.15.010
1 北京軍區總醫院麻醉科 2 天津市胸科醫院心內科
3 河北省巨鹿縣醫院麻醉科
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年6期)2019-10-08 08:55:48
人大建設(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(2017年6期)2017-09-26 11:50:44
學苑創造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國火炬(2010年12期)2010-07-25 13:26:22
中國火炬(2010年8期)2010-07-25 11:34:30
