良性嬰兒癲癇PRRT2基因突變及遺傳特點(diǎn)研究①
2017-08-30 10:02:35劉文婷RakshyaKoirala王菊莉
黑龍江醫(yī)藥科學(xué) 2017年4期
劉文婷,Rakshya Koirala,王菊莉
(1.佳木斯大學(xué)康復(fù)醫(yī)學(xué)院,黑龍江 佳木斯 154003;2.佳木斯大學(xué)臨床醫(yī)學(xué)院,黑龍江 佳木斯 154003;3.佳木斯市中心醫(yī)院癲癇中心,黑龍江 佳木斯 154003)
良性嬰兒癲癇PRRT2基因突變及遺傳特點(diǎn)研究①
劉文婷1,Rakshya Koirala2,王菊莉3
(1.佳木斯大學(xué)康復(fù)醫(yī)學(xué)院,黑龍江 佳木斯 154003;2.佳木斯大學(xué)臨床醫(yī)學(xué)院,黑龍江 佳木斯 154003;3.佳木斯市中心醫(yī)院癲癇中心,黑龍江 佳木斯 154003)
目的:檢測(cè)PRRT2基因在散發(fā)性良性嬰兒癲癇(benign infantile epilepsy,BIE)及家族性良性嬰兒癲癇(benign familial infantile epilepsy,BFIE)患者中的突變情況及探究其遺傳特點(diǎn)。方法:收集在佳木斯市中心醫(yī)院癲癇中心診斷為良性嬰兒癲癇患者,共8例散發(fā)性,一個(gè)BFIE伴陣發(fā)性運(yùn)動(dòng)誘發(fā)性運(yùn)動(dòng)障礙(Paroxysmal Kinesigenic Dyskinesia,PKD)家系共13例,其中4例患病,2例為BFIE,2例為PKD,簽署知情同意書,采集外周靜脈血5mL,通過直接測(cè)序的方法測(cè)取PRRT2基因突變情況,選取10例健康者作為對(duì)照組。采集病史及家族史,對(duì)PRRT2基因突變情況及遺傳特點(diǎn)進(jìn)行分析。結(jié)果:8例散發(fā)患者PRRT2基因無突變;家系中的4例患者均發(fā)現(xiàn)c.640_641insC,符合常染色體顯性遺傳的特點(diǎn)。結(jié)論:PRRT2基因突變可導(dǎo)致BFIE和PKD,且遺傳符合常染色體顯性遺傳特點(diǎn);同一突變點(diǎn)c.640_641insC可導(dǎo)致同一個(gè)家族出現(xiàn)BFIE和PKD,具有遺傳多樣性;PRRT2基因突變可能不是導(dǎo)致散發(fā)BIE的主要原因。
良性嬰兒癲癇;PRRT2;遺傳特點(diǎn)
癲癇(epilepsy) 是一種普通的常見神經(jīng)系統(tǒng)疾病,我國(guó)約患病率3.5‰ ~ 4.8‰[1]。慶幸的是,通過正規(guī)合理的應(yīng)用抗癲癇藥物,約70% 的癲癇患者可以達(dá)到較為滿意的效果[2]。嬰兒期起病的良性局灶性癲癇,預(yù)后好。在2010年,國(guó)際抗癲癇聯(lián)盟國(guó)際抗癲癇聯(lián)盟(International league against epilepsy,ILAE)將此類癲癇分為良性嬰兒癲癇及良性家族性嬰兒癲癇。大量研究表明,癲癇和遺傳因素有關(guān)[3],2011年Chen等[4]在八個(gè)中國(guó)陣發(fā)性運(yùn)動(dòng)誘發(fā)性運(yùn)動(dòng)障礙(Paroxysmal Kinesigenic Dyskinesia,PKD)家族中發(fā)現(xiàn)了富脯氨酸跨膜蛋白2(proline-rich transmenbrane protein 2,PRRT2)突變。此后,一些研究[5]也發(fā)現(xiàn)PRRT2突變出現(xiàn)在BFIE中。本實(shí)驗(yàn)將研究中國(guó)漢族人中散發(fā)性BIE患者及BFIE家系PRRT2基因突變情況。
1 對(duì)象和方法
1.1 對(duì)象入選標(biāo)準(zhǔn)
納入標(biāo)準(zhǔn):①漢族,年齡、性別無要求; ②經(jīng)癲癇科醫(yī)師確診為BIE及BFIE,包括接受藥物治療和未接受藥物治療者。如頭顱CT或者M(jìn)RI及腦電圖均正常;③同意參與本研究并且患者本人或者其監(jiān)護(hù)人簽署知情同意書。
排除標(biāo)準(zhǔn):①患有嚴(yán)重的器官衰竭者、傳染病、血液及內(nèi)分泌系統(tǒng)疾病者;②不愿配合本研究者。
1.2 一般資料
本研究對(duì)象分為散發(fā)性BIE和家族性BIE,其中8例散發(fā)患者,及1個(gè)家系共13例,患病者4例。抽取外周靜脈血5mL,分別為Ⅱ代中的5號(hào)和6號(hào),Ⅲ代中的3號(hào)、4號(hào)和5號(hào)、Ⅳ代中的1號(hào)以及所有散發(fā)患者,進(jìn)行PRRT2基因檢測(cè),同時(shí)選10例健康人作為對(duì)照組。本研究家系中Ⅳ1為先證者,診斷為BFIE,Ⅲ3為先證者舅舅,也診斷為BFIE,先證者母親及外婆,即Ⅲ5及Ⅱ6,均在少年早期患PKD。家系其他成員均健康,家系中Ⅰ代中的男性已去世。家系圖譜如圖1。收集病史及家族史,所有研究對(duì)象均經(jīng)佳木斯中心醫(yī)院癲癇中心診斷為BIE,診斷符合國(guó)際抗癲癇聯(lián)盟(ILAE)的診斷標(biāo)準(zhǔn)。研究對(duì)象全部來自黑龍江省佳木斯市中心醫(yī)院癲癇中心門診及住院部,所有研究對(duì)象均簽署知情同意書。

圖1 BFIE/PKD家系圖譜
1.3 實(shí)驗(yàn)方法
1.3.1 血清DNA提取:采靜周圍脈血5mL,采用Qiagen FlexiGene DNA Kit提取試劑盒提取血清DNA,-20℃保存。
1.3.2 引物設(shè)計(jì):應(yīng)用引物設(shè)計(jì)軟件Primer5.0對(duì)PRRT2基因的3個(gè)外顯子區(qū)域進(jìn)行引物的設(shè)計(jì),設(shè)計(jì)的引物由北京康旭醫(yī)學(xué)檢驗(yàn)所合成。引物序列見表1。
1.3.3 PCR反應(yīng):稀釋引物,根據(jù)引物條件進(jìn)行PCR擴(kuò)增,PCR反應(yīng)體系配制見表2。
1.3.4 DNA測(cè)序:對(duì)所得的PCR產(chǎn)物進(jìn)行1%瓊脂糖凝膠電泳,進(jìn)行鑒定、純化及回收產(chǎn)物,由北京康旭醫(yī)學(xué)檢驗(yàn)所測(cè)序。在BLAST上對(duì)測(cè)序結(jié)果進(jìn)行比對(duì)分析。
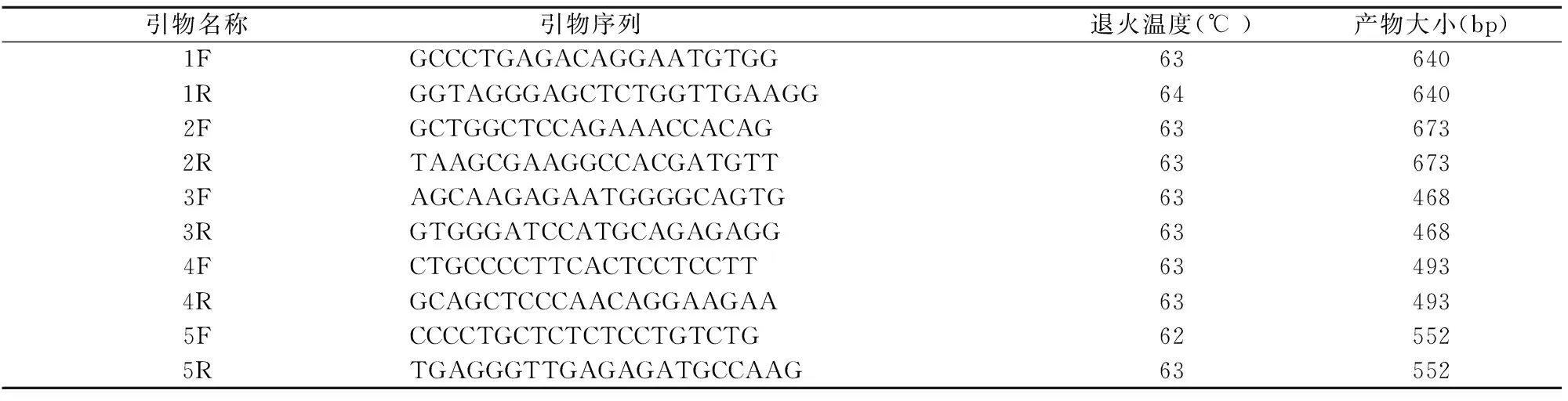
表1 PRRT2引物序列
注:F:正向;R:反向。1F、2F、2R、3F、3R、4F、4R、5R反應(yīng)條件:95℃預(yù)變性10min,94℃ 30s、 63℃ 30s、72℃ 45s 擴(kuò)增34個(gè)循環(huán),72℃延伸5mins,4℃放置。1R反應(yīng)條件:95℃預(yù)變性10min,94℃ 30s、 64℃ 30s、72℃ 45s 擴(kuò)增34個(gè)循環(huán),72℃延伸5mins,4℃放置。5F反應(yīng)條件:95℃預(yù)變性10min,94℃ 30s、 62℃ 30s、72℃ 45s 擴(kuò)增34個(gè)循環(huán),72℃延伸5mins,4℃放置。
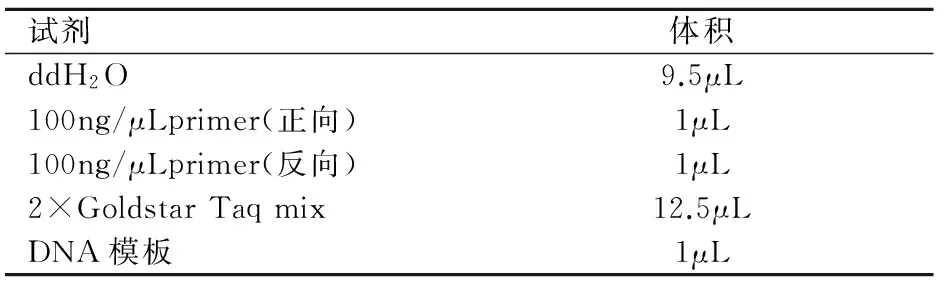
表2 PCR反應(yīng)體系
1.4 統(tǒng)計(jì)學(xué)方法
采用SPSS17.0軟件進(jìn)行分析,由于總樣本量n<40,因此用Fisher確切概率法進(jìn)行對(duì)比,P<0.05有統(tǒng)計(jì)學(xué)意義。
2 結(jié)果
2.1 家系患者PRRT2基因檢測(cè)結(jié)果及分析
該家系中有4位患者,其中兩位為BIE(Ⅳ1、Ⅲ3),兩位為PKD(Ⅱ6、Ⅲ5)。在接受檢測(cè)的成員中此4例患者均檢測(cè)出了PRRT2基因雜合突變,該突變點(diǎn)位于chr16:29825015,為c.640_641insC雜合突變(如圖3)。10例健康對(duì)照組未檢測(cè)出突變(如圖2)。Fisher確切概率法分析得出該家族的患者基因突變頻率有統(tǒng)計(jì)學(xué)意義(P=0.001,P<0.05)見表3。該家系BFIE患者和PKD患者基因突變頻率均有統(tǒng)計(jì)學(xué)意義,見表4、5。說明該基因突變的致病性可信。

表3 家系患者組與健康對(duì)照組PRRT2基因突變頻率分析比較
注:P=0.001,P<0.05。
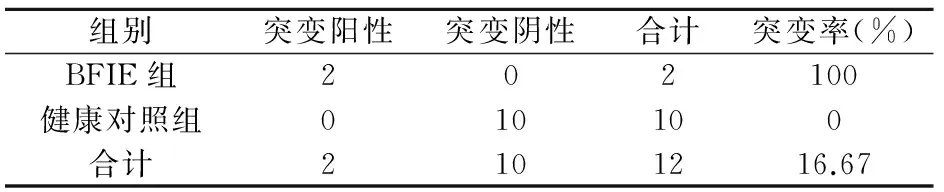
表4 BFIE組與健康對(duì)照組PRRT2基因突變頻率分析比較
注:P=0.015,P<0.05。
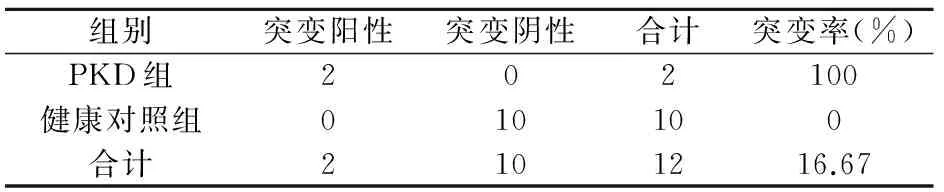
表5 PKD組與健康對(duì)照組PRRT2基因突變頻率分析比較
注:P=0.015,P<0.05。
c.640_641insC為編碼區(qū)第640_641號(hào)核苷酸間插入C的雜合核苷酸變異,該變異導(dǎo)致從第217號(hào)氨基酸Arg開始的氨基酸合成發(fā)生改變,并在改變后的第8個(gè)氨基酸終止(p.Arg217ProfsTer8),為移碼變異,蛋白質(zhì)功能可能因此受到影響。該變異在人群中的發(fā)生頻率極低,從1000Genomes和dbSNP數(shù)據(jù)庫(kù)也可知該變異不屬于多態(tài)性變化。

圖2 正常對(duì)照基因測(cè)序圖
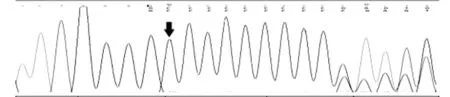
圖3 突變基因測(cè)序圖
2.2 散發(fā)BIE患者基因檢測(cè)結(jié)果
8例散發(fā)患者均未檢測(cè)出PRRT2基因突變,突變頻率為0,與健康對(duì)照組突變頻率一致。
2.3 家系遺傳學(xué)特征
由圖1可知,該家系連續(xù)3代人發(fā)病,Ⅱ6、Ⅲ3、Ⅲ5、Ⅳ1都具有PRRT2基因雜合突變,且都患病。Ⅱ6與Ⅲ5和Ⅳ1為直系親屬,Ⅱ6與Ⅲ3為直系親屬,符合孟德爾常染色體顯性遺傳規(guī)律。由于Ⅰ代中的男性已去世,所以無法追蹤此突變遺傳是否伴有不全外顯。該家族4例患者同一個(gè)突變點(diǎn)卻患有兩種病癥,且都只是單純患有BIE或者單純患PKD,沒有BIE轉(zhuǎn)換為PKD,也沒有PKD伴有BIE。說明PRRT2基因突變具有遺傳多效性。
3 討論
Fukuyama[6]在1963年第一次報(bào)告嬰兒良性癲癇后,隨著對(duì)嬰兒良性癲癇的深入研究,其診斷標(biāo)準(zhǔn)也不斷地被改進(jìn),我國(guó)一般采用劉等[7]提出的診斷標(biāo)準(zhǔn), 本研究收集的患者對(duì)單一抗癲癇藥物反應(yīng)好,預(yù)后好,符合診斷標(biāo)準(zhǔn)。本研究收錄的8例患者均未檢測(cè)出PRRT2基因突變,雖然有報(bào)道[8]散發(fā)性BIE患者PRRT2基因檢測(cè)出突變,突變率為28.5%,而日本一關(guān)于散發(fā)性BIE的研究也未發(fā)現(xiàn)PRRT2基因突變[9]。表明PRRT2可能不是散發(fā)性嬰兒良性癲癇的主要致病基因,或者提示中國(guó)漢族人種的散發(fā)的良性嬰兒癲癇患者PRRT2基因突變率低,亦可能與本病散發(fā)病例例數(shù)偏少有關(guān),或可能存在末端調(diào)控序列突變或者外顯子丟失等機(jī)制,不能被常規(guī)檢測(cè)方法所檢測(cè)出。因此,對(duì)于散發(fā)性良性嬰兒癲癇的主要致病基因的研究應(yīng)該轉(zhuǎn)向其他基因,如KCNQ2基因和KCNQ3基因等。
c.640_641insC為編碼區(qū)第640_641號(hào)核苷酸間插入C的雜合核苷酸變異,有報(bào)告將c.640_641insC寫成c.649dupC(p.R217PfsX8)或者c.649_650insC[9],這三種不同的寫法都表示該變異導(dǎo)致從第217號(hào)氨基酸Arg開始的氨基酸合成發(fā)生改變,并在改變后的第8個(gè)氨基酸終止(p.Arg217ProfsTer8),為移碼變異,蛋白質(zhì)功能可能因此受到影響。該變異在正常人群中的發(fā)生頻率極低,從1000Genomes和dbSNP數(shù)據(jù)庫(kù)可知該變異不屬于多態(tài)性變化。
近幾年來,隨著國(guó)內(nèi)外對(duì)PRRT2研究越來越廣泛和深入,一些研究報(bào)告在其他疾病中也發(fā)現(xiàn)了PRRT2基因的突變點(diǎn)c.649_650insC[10,11]。韓國(guó)報(bào)道一患有PKD伴BIC且在發(fā)作期發(fā)現(xiàn)中央顳區(qū)棘波(centrotemporal spikes,CTS)的10歲女孩,在基因檢測(cè)中發(fā)現(xiàn)c.649_650insC[10]。 有研究報(bào)道一例患有發(fā)作性過度運(yùn)動(dòng)障礙(paroxysmal hypnogenic dyskinesia,PHD)的患者也發(fā)現(xiàn)該突變位點(diǎn)c.649_650insC[11]。以上研究進(jìn)一步證實(shí)了PRRT2基因突變具有遺傳多效性。 一個(gè)基因甚至一個(gè)基因位點(diǎn)突變可導(dǎo)致如此多病癥,因此有人推測(cè)上述一系列發(fā)作性神經(jīng)系統(tǒng)疾病在遺傳學(xué)方面可能屬于同一種疾病,只是臨床表現(xiàn)不同而已,由此提出PRRT2相關(guān)疾病這一概念[12]。最近一研究在一陣發(fā)性運(yùn)動(dòng)源性運(yùn)動(dòng)障礙伴嬰兒驚厥(paroxysmal kinesigenic dyskinesia with infantile convulsion,PKD/IC)的患兒身上發(fā)現(xiàn)了PRRT2內(nèi)含子突變,其母親患有PKD,因此該研究者提出此內(nèi)含子突變可能是導(dǎo)致這個(gè)家族患病的原因,由于移碼或者提前終止密碼子而使PRRT2蛋白功能可能紊亂[13]。由此提醒我們,在研究PRRT2致病的機(jī)理時(shí),也應(yīng)該注意其內(nèi)含子的作用。本研究由于條件有限未進(jìn)行內(nèi)含子的檢測(cè)。
同樣日本一研究也對(duì)一個(gè)BFIE/PKD家系進(jìn)行了PRRT2基因檢測(cè),與本研究結(jié)果一致,BFIE及PKD患者檢測(cè)出c.649_650insC突變,而j家族成員中健康者未檢測(cè)出該基因突變[9]。關(guān)于PRRT2基因突變引起的一系列疾病的研究發(fā)現(xiàn),有超過80%的家族性BIE有PRRT2基因突變[14]。本研究該家系如此高突變頻率也在一定程度上佐證了這一觀點(diǎn)。綜上所述,在對(duì)家族性BIE患者做PRRT2基因檢測(cè)是有必要的,而對(duì)散發(fā)性BIE患者做PRRT2基因檢測(cè)的必要性還需進(jìn)一步研究探討。
[1]和梅,徐金銘,王欣. 葉酸聯(lián)用維生素B12對(duì)丙戊酸鈉治療的癲癇患者血同型半胱氨酸水平的影響[J]. 黑龍江醫(yī)藥科學(xué),2016,39(2):53-54
[2]任會(huì)菊,喬洪朝. 26例癲癇治療失敗的原因分析[J]. 黑龍江醫(yī)藥科學(xué),2012,35(5):79
[3]李欣宇,左松林,張昭,等. SCN9A基因多態(tài)與兒童良性癲癇相關(guān)性[J]. 黑龍江醫(yī)藥科學(xué),2015,38(6):24-26
[4]Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia[J]. Nat Genet,2011,43:1252-1255
[5]Schubert J, Paravidino R, Becker F, et al. PRRT2 Mutations are the major cause of benign familial infantile seizures[J]. Human Mutation,2012,33(10):1439-1443
[6]Berg AT, Berkovic SF, Brodie MJ, et al.Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009[J]. Epilepsia,2010,51:676-685
[7]劉曉燕,姜玉武,吳懼,等. 嬰兒良性癲癇的臨床觀察和遠(yuǎn)期隨訪研究[J]. 中華兒科雜志, 2003, 41(1):14-16
[8]Specchio N, Terracciano A, Trivisano M, et al. PRRT2 is mutated in familial and non-familial benign infantile seizures[J]. Eur J Paediatr Neurol,2013,17(1):77-81
[9]Okumura A,Shimojima K,Kubota T,et al.PRRT2 mutation in Japanese children with benign infantile epilepsy[J]. Brain & Development ,2013,35(7):641-646
[10]Seo SY,You SJ.Paroxysmal kinesigenic dyskinesia in a patient with a PRRT2 mutation and centrotemporal spike discharges on electroencephalogram: case report of a 10-year-old girl[J]. Korean J Pediatr,2016,59(1):157-160
[11]Liu XR,Huang D,Wang J,et al.Paroxysmal hypnogenic dyskinesia is associated with mutations in the PRRT2 gene[J]. Neurol Genet,2016,2(2):66
[12]Becler F,Schubert J,Stroano P,et al.PRRT2-relateddisorders: further PKD and ICCA cases and review of the literature[J]. J Neurol, 2013,52(12):980-987
[13]Weber A,Kreth J,Müller U. Intronic PRRT2 mutation generates novel splice acceptor site and causes paroxysmal kinesigenic dyskinesia with infantile convulsions(PKD/IC) in a three generation family[J]. BMC Med Genet,2016,17(16):281
[14]Heron SE, Dibbens LM. Role of PRRT2 in common paroxysmal neurological disorders:a gene with remarkable leiotropy[J]. J Med Genet,2013,50:133-139
Highlights: Objective: To detect the PRRT2 gene mutation and explore genetic characteristics in patients with sporadic benign infantile epilepsy (BIE) or benign familial infantile epilepsy (BFIE). Methods: 8 cases of sporadic BIE diagnosed by Jiamusi Central Hospital were collected. A family of 13 people, 2 person had BFIE and 2 person had paroxysmal kinesigenic dyskinesia (PKD) were used in this study. All participants were signed a written informed consent. 5ml peripheral venous blood was collected from participants. PRRT2 gene mutation sequencing were directly measured. 10 healthy subjects were selected as control group. History and family history were collected to analyze the PRRT2 gene mutation and genetic characteristics. Results: There was no PRRT2 gene mutation in 8 sporadic patients. C.640_641insC was found in all 4 patients of the family. Conclusion: PRRT2 gene mutation can lead to BFIE and PKD, and satisfied the genetic characteristics of autosomal dominant inheritance; the same mutation point c.640_641insC can lead to BFIE and PKD in a family with genetic diversity; PRRT2 gene mutation may not be the main cause of sporadic BIE.
Study on PRRT2 gene mutation and genetic characteristics of benign infantile epilepsy
LIUWen-ting1,RakshyaKoirala2,WANGJu-li3
(1. College of Rehabilitation Medicine in Jiamusi University, Jiamusi 154003,China; 2. College of Clinical Medicine in Jiamusi University, Jiamusi 154003,China; 3. Epilepsy Center of Jiamusi Central Hospital, Jiamusi 154003,China)
BIE; PRRT2;genetic characteristics
佳木斯大學(xué)研究生科技創(chuàng)新項(xiàng)目,編號(hào):YM2016_090。
劉文婷(1989~)女,陜西渭南人,在讀碩士研究生。
王菊莉(1964~)女,黑龍江佳木斯人,博士,主任醫(yī)師,教授,碩士研究生導(dǎo)師。E-mail:wjll98981@163.com。
R722.19
B
1008-0104(2017)04-0032-04
2016-12-12)
猜你喜歡
英語(yǔ)世界(2023年6期)2023-06-30 06:29:10
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中國(guó)民間療法(2021年5期)2021-06-09 09:21:04
中國(guó)生殖健康(2020年2期)2021-01-18 02:51:26
小學(xué)生導(dǎo)刊(2018年13期)2018-06-29 03:49:00
飲食科學(xué)(2017年5期)2017-05-20 17:11:53
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
西南軍醫(yī)(2015年4期)2015-01-23 01:19:30
