1例疑診Leigh綜合征患兒及其父母線粒體相關基因突變觀察
2017-08-09 01:27:44李亞麗孫艷美張寧王方娜高健
山東醫藥 2017年27期
關鍵詞:基因突變
李亞麗,孫艷美,張寧,王方娜,高健
(河北省人民醫院,石家莊050071)
?
1例疑診Leigh綜合征患兒及其父母線粒體相關基因突變觀察
李亞麗,孫艷美,張寧,王方娜,高健
(河北省人民醫院,石家莊050071)
目的 觀察并分析1例疑診Leigh綜合征患兒及其父母的線粒體相關基因突變情況。方法 發育落后合并肌張力異常、疑診Leigh綜合征的患兒1例,3歲,患兒就診時其母孕14周。采集患兒及其父母的外周血2 mL,提取血液基因組DNA,采用高通量測序分析檢測患兒線粒體病相關基因,并觀察其父母基因突變情況。根據檢測出的患兒線粒體相關基因突變位點,在患兒母親孕18周時抽取羊水,采用PCR法檢測胎兒是否存在相同突變位點,分析胎兒患病的風險。結果 測得患兒過量位點蛋白1(SURF1)基因有兩個突變位點c.655G>T(p.E219X)和c.324-1G>C,c.655G>T來源于母親、c.324-1G>C來源于父親,為復合雜合突變,確診患兒為疑診Leigh綜合征。患兒母親羊水PCR檢測分析發現胎兒為c.324-1G>C突變,無c.655G>T突變,為雜合突變攜帶者,胎兒出生后隨訪至1歲,孩子身體健康,能平穩獨自行走。結論 發現Leigh綜合征患兒SURF1基因的兩個突變位點c.655G>T(p.E219X)、c.324-1G>C,是患兒發育落后、肌張力異常的致病基因;患兒父母均為雜合基因突變攜帶者,遺傳方式是常染色體隱性遺傳,母親每次受孕其胎兒患病概率為25%。
線粒體肌病;Leigh綜合征;過量位點蛋白1基因;基因突變;常染色體隱性遺傳
線粒體是真核細胞中的一種細胞器,它是細胞內氧化磷酸化和合成三磷酸腺苷(ATP)的主要場所,為細胞95%的活動提供所需能量,并且它還參與細胞分化、細胞信息傳遞和細胞凋亡等過程,有調控細胞生長和細胞周期的能力。線粒體有自身的遺傳物質和遺傳體系,線粒體基因約編碼人類2%的蛋白質[1]。線粒體肌病是由于線粒體結構和功能異常造成細胞呼吸鏈及能量代謝障礙的臨床綜合征,當其伴有中樞神經系統癥狀時稱為線粒體腦肌病。線粒體肌病較常見的一種臨床類型是Leigh綜合征(LS),多發生于嬰幼兒或兒童期,起病較隱匿,呈慢性進行性的肌張力倒退、伴隨運動和智力發育遲緩,部分患者可出現呼吸衰竭、運動障礙而危及生命。早期診斷和治療LS非常重要。該病臨床表現多樣,單純依靠臨床表現和一般實驗室檢查很難準確診斷,目前診斷該病的金標準是基因診斷[2~4]。本研究回顧分析了對1例臨床疑似LS患兒的分子遺傳學研究過程,發現了過量位點蛋白1(SURF1)基因的兩個新的突變位點,明確了診斷和病因,并對其母孕期進行了產前診斷,現報告如下。
1 資料與方法
1.1 臨床資料 疑似LS患兒1例,女,3歲,發育落后。生后9個月始能獨坐,1歲3個月可以扶走,扶走時雙下肢抬起困難,至今不能獨自站立和行走;僅能簡單發音“爸爸、媽媽”;1歲3個月運動倒退,四肢無力,嘔吐、喂養困難,易哭鬧、睡眠差,血乳酸3.0 mm/L;頭MRI檢查示雙側基底節區及腦干異常信號,部分病變彌散受限。根據患兒臨床癥狀和檢驗結果疑診LS。患兒父母同齡(25歲),身體均健康,否認家族中有遺傳病史。患兒母親25歲,23歲足月順產此患兒,為第1次孕產,患兒生后Apgar評分正常。患兒母親現孕14周,為避免再次生育此類患兒,要求行產前診斷。
1.2 線粒體相關基因突變觀察 采集患兒及其父母的外周血2 mL(EDTA抗凝),按照血液基因組DNA提取試劑盒說明書提取基因組DNA(天根試劑盒)。以電泳和NanoDrop超微量分光光度計檢測DNA的質量和濃度。提取1 500 ng的基因組DNA,超聲打斷成平均300 bp的長度,進行文庫構建;連接8 bp的index標簽和接頭到打斷的DNA上;連接好接頭的片段化DNA與內分泌疾病捕獲芯片進行雜交捕獲;捕獲后的文庫質量和濃度通過片段分析和定量PCR進行評估。上illumina HiSeq X ten平臺測序,基因組中佝僂病相關基因的測序數據量在2 G左右,平均測序深度在100 X。測序數據通過NextGENe軟件進行數據過濾和比對分析。篩選的突變點在數據庫(dbSNP、ExAC和1000 genome projects)中比對來確定人群攜帶率,進一步判斷其致病性;通過SIFT、Polyphen-2 and mutation taster等軟件進行突變點保守性及致病性分析,預測其致病性。對疑似致病的突變位點,通過一代測序進行驗證,測序結果借助Mutation Surveyor v4.0軟件與參考序列進行比對。
1.3 患兒母親羊水細胞中線粒體相關基因突變觀察 患兒母親于孕18周時抽取羊水,采用PCR法檢測胎兒是否存在相同突變位點,進行產前診斷。于產后1年對胎兒出生后的情況進行隨訪。
2 結果
測得患兒SURF1基因有兩個突變位點c.655G>T(p.E219X)和c.324-1G>C,c.655G>T來源于母親、c.324-1G>C來源于父親,為復合雜合突變(見圖1、2),確診患兒為LS。患兒母親羊水PCR檢測分析發現胎兒存在c.324-1G>C突變,未發現c.655G>T突變,為雜合突變攜帶者(見圖1、2)。患兒母親足月順產一男嬰,現已1歲,身體健康,能平穩獨自行走。
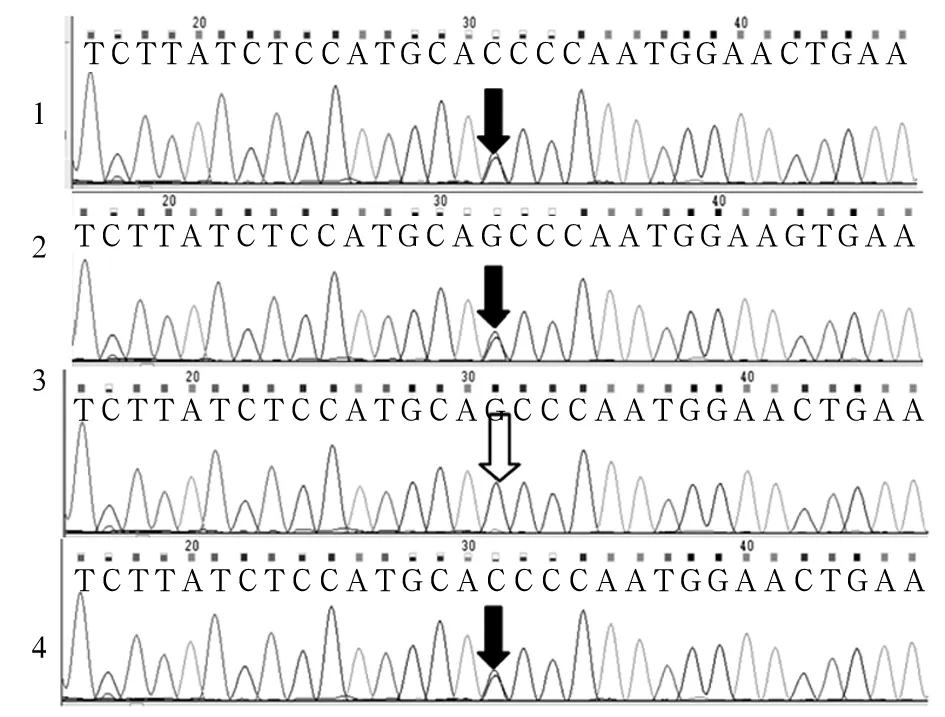
注:1為患兒樣本;2為患兒父親樣本;3為患兒母親樣本;4為羊水細胞樣本;黑色箭頭示SURF1基因突變位點c.655G>T(p.E219X);白色箭頭處SURF1基因不存在突變位點c.655G>T(p.E219X)。
圖1 SURF1基因突變位點c.655G>T(p.E219X)
3 討論
LS最早由英國學者[5]發現并描述其神經病理學特征。LS是多種酶缺陷導致的線粒體呼吸鏈氧化磷酸化障礙,引起退行性中樞神經系統損害,其特征是腦干、基底節和脊髓后柱區有對稱性局灶壞死,故又稱為亞急性壞死性腦脊髓病[6,7]。LS病因復雜,臨床發病率約1/40 000。LS按患者發病年齡可分為三種類型,即新生兒型、經典嬰兒型、青少年型。其中以嬰兒型最常見,臨床表現不一,包括運動和智力低下(包括舞蹈癥)、肌張力減退、痙攣、小腦共濟失調等,發病后2年內病死率高[8~10]。LS患者血清或腦脊液中乳酸含量增高[11~12]。祝小芬等[13]研究認為靜息時乳酸升高、運動后即刻超過正常值3倍可作為擬診LS的參考。LS頭部MRI檢查特征性表現是基底節尤其殼核區可見多灶性、雙側對稱性的腦軟化灶。病理學研究發現LS患者丘腦、基底節、腦室、大腦導水管周圍和脊髓后柱區有對稱性局灶壞死,呈海綿狀囊樣空腔,并伴有脫髓鞘改變、血管增生和神經膠質增生[14]。LS具有復雜的臨床表現和基因調控機制,為嚴重的致死性遺傳病,且目前無有效治療措施,故產前診斷是一項重要預防措施。
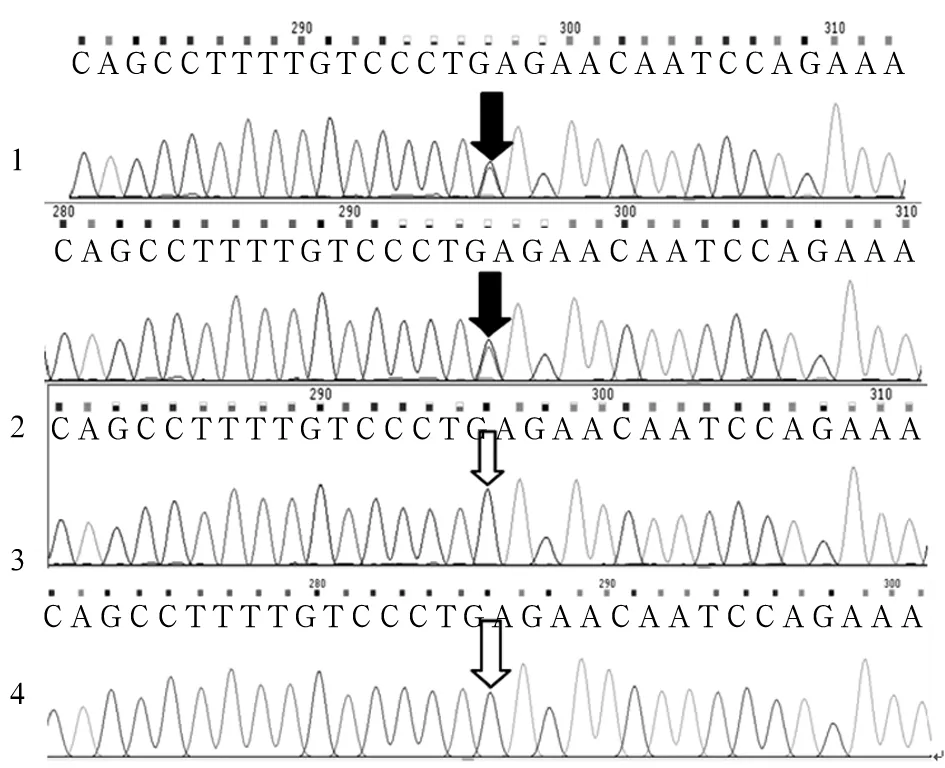
注:1為患兒樣本;2為患兒父親樣本;3為患兒母親樣本;4為羊水細胞樣本;黑色箭頭示SURF1基因存在突變位點c.324-1G>C,c.655G>T;白色箭頭處SURF1基因不存在突變位點c.324-1G>C,c.655G>T。
圖2 SURF1基因突變位點c.324-1G>C,c.655G>T
LS主要有3種遺傳方式:線粒體遺傳(又稱母系遺傳),由線粒體基因突變引起;常染色體隱性遺傳;X-連鎖遺傳。后兩種遺傳方式是核基因突變所致,其中常染色體隱性遺傳最為常見,約占60%。目前發現約10種類型核基因突變引起線粒體病,包括呼吸鏈復合物亞基及組裝因子突變,維持線粒體基因穩定性的突變,線粒體蛋白組合裝置缺陷,線粒體脂質環境缺陷,線粒體運輸裝置缺陷,輔酶Q10缺乏,線粒體網絡動態學缺陷,凋亡缺陷,丙酮酸脫氫酶缺陷和線粒體鐵代謝障礙[3]。呼吸鏈復合物亞基及組裝因子突變基因達51個[15]。研究[16,17]發現常染色體隱性遺傳與4種呼吸鏈酶缺陷有關,即復合物Ⅰ(NADH還原酶)、復合物Ⅱ(琥珀酸脫氫酶)、復合物Ⅳ(COX)、復合物Ⅴ(ATP合成酶)。其中最為常見是復合物Ⅳ的缺陷。COX復合物由13個多肽亞單位組成,其中10個由核基因編碼(SURF1、SCO1、SCO2、COX10、COX15、COX20、LRPPRC、FASTKD2、TACO1、PET100等),其余3個由線粒體基因編碼[18,19]。LS最常見的致病基因是SURF1,位于9q34.2,有9個外顯子,全長4 695 bp。SURF1基因編碼的蛋白含有300個氨基酸殘基(AA),有2個跨膜結構域(61~79AA、274~290AA)。SURF1蛋白在COX復合物的裝配前期中發揮十分重要的作用。迄今國外報道已發現40多例LS患者存在SURF1基因突變[20];國內共報道了6例,1例為622delA和653 -654delCT缺失,5例為外顯子7的604位G>C雜合錯義突變。
本研究中患兒發育落后,不能獨走,扶走時雙下肢抬起困難,2歲后活動能力有倒退;血乳酸3.0 mmol/L;頭部MRI檢查示雙側基底節區及腦干異常信號,部分病變彌散受限,符合LS的臨床特征,擬診斷為LS。進行二代測序篩查基因發現患兒SURF1基因有c.655G>T(p.E219X)及c.324-1G>C復合雜合突變,與PubMed文獻數據庫、美國生物技術信息中心(NCBI)所屬的SNP數據庫、人類基因突變數據庫(HGMD)中已公布的SURF1基因突變位點相比,這兩個突變均未見報道。上述兩個突變中,前者為無義突變,后者為剪切突變,理論上均具有致病性。故我們認為這兩種缺失突變為新的致病突變。傳遞分析表明,患兒母親存在SURF1基因c.655G>T雜合突變,父親有SURF1基因c.324-1G>C雜合突變,明確了診斷和致病基因,不僅為患兒的治療提供了準確依據,而且根據患兒致病SURF1基因及其父母基因比對,發現患兒父母親均為致病基因攜帶者,遺傳方式是常染色體隱性遺傳,故患兒父母再次生育時,其子女患病概率為25%,需進行產前診斷。本例患兒母親在孕18周時進行羊水細胞PCR檢測,發現胎兒有c.324-1G>C突變,沒有c.655G>T突變,為雜合突變攜帶者,理論上不具致病性,故保留此胎兒,產后隨訪至1歲,孩子未出現異常。
[1] 李欣,曹翔,汪麟,等.疑難病例分析:線粒體腦病1例[J].齊齊哈爾醫學院學報,2015,36(13):2027-2028.
[2] Graham BH. Diagnostic challenges of mitochondrial disorders:complexities of two genomes[J]. Methods Mol Biol, 2012,837:35-46.
[3] 劉志梅,方方.核基因突變導致兒童線粒體病的分子遺傳學進展[J].中國循證兒科雜志,2015,10(6):470-474.
[4] Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diagnostic applications in children[J]. Neurology, 2006,67(10):1823-1826.
[5] Leigh D. Subacute necrotizing encephalomyelopathy in an infant[J]. Neurol Neurosurg Ychiatry, 1951,14:216-221.
[6] 劉雪梅,魏曉晶,蘇飛飛,等.Leigh綜合征1例報告[J].中風與神經疾病雜志,2017,34(2):177-178.
[7] DiMauro S, Schon EA. Mitochondrial disorders in the nervous system[J]. Annu Rev Neurosci, 2008,31(10):91-123.
[8] Scheffer RC, Smout AJ. Tachyduodenia in mitochondrial neurogastrointestinal encephalomyopathy[J]. Neurogastroenterol Motil, 2011,23(5):408-410.
[9] 常凱杰,馬明明,張曉輝,等.肢帶型線粒體肌病一個家系的臨床和病理特點研究[J].國際神經病學神經外科學雜志,2016,43(1):22-26.
[10] 王麗輝,鄭華城,楊花芳,等.兒童Leigh綜合征4例臨床分析[J].臨床兒科雜志,2016,34(2):111-114.
[11] Ruhoy IS, Saneto RP. The genetics of Leigh syndrome and its implications for clinical practice and risk management[J]. Appl Clin Genet, 2014,7(12):221-234.
[12] 孟慶林,江煒煒.線粒體腦肌病伴高乳酸血癥和卒中樣發作綜合征的臨床特點分析[J].臨床神經病學雜志,2016,29(4):274-277.
[13] 祝小芬,丁冀,朱筱琦,等.線粒體肌病臨床病理學特征及簡易乳酸運動試驗的篩選價值[J].中國現代神經疾病雜志,2016,16(12):859-864.
[14] 盧萬協.嬰幼兒Leigh綜合征的MRI診斷探討分析[J].河南預防醫學雜志,2009,20(5):402-403.
[15] Chinnery PF, Hudson G. Mitochondrial genetics[J]. Br Med Bull, 2013,106(5):135-159.
[16] Haack TB, Haberberger B, Frisch EM, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing[J]. Med Genet, 2012,49(4):277-283.
[17] Nogueira C, Barros J, Sa MJ, et al. Novel TTC19 mutation in a family with severe psychiatric manifestations and complex III deficiency[J]. Neurogenetics, 2013,14(2):153-160.
[18] Bohm M, Pronicka E, Karczmarewicz E, et al. Retrospective, Multicentric Study of 180 Children with Cytochrome c Oxidase Deficiency[J]. Pediatr Res, 2006,59(4):21-26.
[19] Szklarczyk R, Wanschers BF, Nijtmans LG, et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia[J]. Hum Mol Genet, 2013,22(4):656-667.
[20] Zhang Y, Sun F, Kong QP, et al. Mutation analysis on nuclear gene and mitochondrial gene for Chinese patients with Leigh syndrome[J]. Clin Pediatr, 2008,26(12):1021-1025.
10.3969/j.issn.1002-266X.2017.27.028
R394
B
1002-266X(2017)27-0090-03
2017-03-05)
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
