脂多糖誘導巨噬細胞中核受體協同抑制因子啟動子甲基化對炎癥因子的調控作用
2017-05-15 03:35:54郭小莉劉霞雷傳江王關嵩王建春
中華肺部疾病雜志(電子版) 2017年2期
郭小莉 劉霞 雷傳江 王關嵩 王建春
?
·論著·
脂多糖誘導巨噬細胞中核受體協同抑制因子啟動子甲基化對炎癥因子的調控作用
郭小莉 劉霞 雷傳江 王關嵩 王建春
目的探討核受體協同抑制因子(NCOR)在脂多糖(LPS)誘導巨噬細胞炎癥反應中的作用及其調控機制。方法1 μg/ml的LPS分別處理小鼠巨噬細胞RAW264.7 24 h和48 h,應用Western blot和Real time-PCR檢測NCOR的表達水平以及腫瘤壞死因子-α(TNF-α)、白細胞介素-6(IL-6) mRNA水平,熒光素酶報告基因檢測核因子-κB (NF-κB)的啟動子活性。LPS處理細胞48 h后,應用MSP檢測NCOR啟動子是否發生甲基化以及Western blot檢測DNMT3b的表達變化。Real time-PCR檢測5′-aza和LPS聯合處理細胞后NCOR mRNA的表達水平;轉染DNMT3b siRNA后,分別應用Western blot和Real time-PCR檢測DNMT3b的表達水平,以及DNMT3b siRNA和LPS聯合作用下NCOR、TNF-α、IL-6的表達水平和NF-κB的啟動子活性。結果LPS干預細胞24、48 h后,NCOR蛋白和mRNA表達顯著下調(P<0.05),而TNF-α、IL-6 mRNA表達水平、DNMT3b 蛋白的表達水平以及NF-κB的啟動子活性顯著上升(P<0.05)。MSP檢測說明LPS可介導NCOR的啟動子甲基化。用5′-aza和LPS聯合處理細胞后NCOR mRNA水平較LPS組有顯著上升(P<0.05)。采用DNMT3b siRNA可顯著下調DNMT3b 蛋白和mRNA水平,并可部分逆轉LPS介導的抑制NCOR表達的效應,抑制TNF-α、IL-6的表達水平和NF-κB的啟動子活性(P<0.05)。結論NCOR啟動子的甲基化是LPS介導巨噬細胞炎癥反應發生、發展的關鍵步驟,其可作為治療ALI/ARDS的潛在靶點。
核受體協同抑制因子; DNA 甲基化; 核轉錄因子κB; 炎癥因子
急性肺損傷(acute lung injury, ALI)和急性呼吸窘迫綜合征(acute respiratory distress syndrome, ARDS)嚴重威脅著人民的生命安全[1-3]。盡管新型藥物及機械通氣等措施得到廣泛的應用,但是ALI/ARDS的病死率仍居高不下[4]。嚴重的炎癥反應如膿毒血癥是引起ALI/ARDS主要原因之一[5]。而在這個過程中,受到內毒素刺激的肺泡巨噬細胞通過大量釋放炎癥介質,在肺組織損傷中發揮著關鍵的作用[5-6]。因此,深入探究肺巨噬細胞在炎癥反應中的分子機制,可為ALI/ARDS的防治提供新的防治策略和靶點。
核受體協同抑制因子(nuclear receptor corepressor, NCOR)是一種核轉錄抑制因子,其通過調控多種轉錄因子發揮作用。Hong等[7]研究發現下調NCOR表達可通過促進過氧化物酶體增生物激活受體γ(peroxisome proliferator-activated receptor gamma, PPARγ)的表達,進而發揮負性調節間充質干細胞向脂肪細胞的分化。其他研究也表明NCOR在調控腫瘤的相關生物學特性方面發揮著重要的作用[8-9]。而在炎癥反應中,通過抑制NCOR的表達,可促進炎癥因子釋放的作用,但是否存在其他機制尚不清楚[10]。
DNA甲基化是一種常見的基因修飾途徑之一。其主要是在DNA甲基轉移酶(DNA methyltransferases, DNMTs)的催化下,完成對染色質結構、DNA構象、DNA穩定性及DNA與蛋白質相互作用方式的改變,控制基因表達,進而引起疾病的發生、發展[11-14]。既往研究表明在ALI大鼠模型中上調DNMT3b、DNMT1等,可引起PPAR γ等基因的啟動子甲基化,進而在ALI的進展中發揮著重要作用[15-16]。本研究旨在探討LPS是否可誘導NCOR的甲基化,及其在NCOR甲基化中的機制,及在巨噬細胞介導的炎癥反應中的作用。
材料和方法
一、實驗材料
DMEM高糖培養基和胎牛血清(無內毒素)購自GIBCO公司;脂多糖(lipopolysaccharide, LPS)和5-氮胞苷2′-脫氧胞苷(5-Aza-2′-deoxycytidine,5′-aza)和TRIzol reagent購自sigma公司;兔源NCOR單克隆抗體、兔源DNMT3b單克隆抗體、兔源GAPDH多克隆抗體以及山羊抗兔HRP-Ig(H+L)二抗購自江蘇睿瀛生物科技有限公司;逆轉錄試劑盒購自Thermo fisher公司;定量PCR mix購自羅氏公司;DNA提取試劑盒、DNA重亞硫酸鹽轉化試劑盒和甲基化特異性PCR試劑盒均購自天根生化科技有限公司。pNF-κB-luc報告基因質粒、G418和熒光素酶報告基因檢測試劑盒購自江蘇碧云天生物技術有限公司。DNMT3b siRNA和陰性對照(negative control, NC)由廣州銳博生物科技有限公司設計和合成;轉染試劑lipofiter購自漢恒(上海)生物科技有限公司。
二、研究方法
1. 小鼠巨噬細胞RAW264.7的培養:小鼠巨噬細胞RAW264.7由第三軍醫大學生物測試中心提供。RAW264.7用DMEM+10%FBS的培養基進行培養。當細胞融合度達到90%時使用細胞刮刀將細胞刮下來,離心后重新進行傳代。細胞傳代后到達對數生長期進行相關實驗。
2. LPS處理RAW264.7細胞:使用1 μg/ml的LPS分別處理對數生長期的RAW264.7細胞0 h、24 h和48 h,然后進行后續試驗[16]。
3. 5′-aza處理RAW264.7細胞:使用1 μM的5′-aza分別處理對數生長期的RAW264.7細胞48 h,然后進行后續試驗。
4. pNF-κB-luc報告基因質粒的穩定轉染:將消化下來的RAW264.7細胞傳代到10 cm2培養皿中,培養12 h使細胞融合度達到80%。棄去培養基加入10 ml的DMEM。將24 μg pNF-κB-luc質粒加入1 ml DMEM中,充分混勻。再取一個EP管,加入1 ml的DMEM后,再加入72 μl lipofiter,充分混勻。將上述兩管靜置5 min后,混合在一起,充分混勻后再靜置15 min。將上述混合液均勻滴入細胞培養基中,培養6 h后換成還有DMEM+10%FBS的完全培養基繼續培養48 h。之后要用1 mg/ml的G418持續篩選1個月,獲得穩定表達NF-κB-luc-RAW264.7細胞。
5. 甲基化特異性PCR(methylating-specific PCR, MSP):將處理后的RAW264.7細胞用PBS洗滌1次后,用細胞刮刀將細胞刮下,離心。用DNA提取試劑盒提取細胞的DNA,使用DNA重亞硫酸鹽轉化試劑盒將DNA樣品中未甲基化胞嘧啶轉化為尿嘧啶。非甲基化引物(unmethylation primer):上游:5′-TTGTTTGTGTTATTTATGTTTATGA -3′,下游: 5′-CAATACTTACTACTACCCACATCAAT-3′。甲基化引物(methylation primer): 上游:5′-TTGTTTGTGTTATTTGTGTTTACGA-3′,下游: 5′-ATACTTACTACTACCCGCGTCGAT-3′,見表1。將上述樣本就行MSP,擴增條件:95 ℃ 3 min預變性,95 ℃ 20 s,60 ℃ 20 s,72 ℃ 20 s進行35個循環,再以72 ℃ 5 min進行延伸。將上述產物進行瓊脂糖電泳。
6. DNMT3b siRNA轉染RAW264.7細胞:將3條DNMT3b siRNA和對照組 (NC)用RNase-free H2O 配制成20 nM的儲存液后,分裝、凍存備用。使用終濃度為50 pM DNMT3b siRNA轉染細胞的簡要操作如下:①將5×104/孔 RAW264.7接種到24孔板中,培養12 h;②2.5 μl 濃度為20 nM的DNMT3b siRNA加入到50 μl DMEM中,輕輕混勻;③2.4 μl的lipofiter 加入到50 μl DMEM中,輕輕混勻;④上述兩組混懸液靜置5 min后,再將它們混合在一起,充分混勻,靜置15 min;⑤將100 μL混合試劑加入含有細胞的孔板中,輕輕混勻,DNMT3b siRNA的轉染終濃度為50 pM;⑥將培養板置于37 ℃、5% CO2、飽和濕度下的培養箱中培養48 h后進行PCR和Western blot等檢測。NC的轉染同前。
7. 熒光素酶活性的檢測:將作為內參pRL-TK質粒和DNMT3b siRNA/NC 共同轉染穩定表達NF-κB-luc的RAW264.7細胞24 h后,再用1 μg/ml LPS干預24 h。將細胞用PBS洗滌1~2次,然后加入200 μl報告基因細胞裂解液,充分裂解細胞后,按照說明書加入溶解熒光素酶檢測試劑,孵育后并進行檢測。
8. Real time-PCR檢測NCOR、DNMT3b、IL-6和TNF-α mRNA的表達:收集細胞后,用TRIzol提取總RNA,并逆轉錄為cDNA. cDNA采用BIO-RAD CFX96系統進行實時熒光定量PCR檢測NCOR、DNMT3b、IL-6和TNF-α mRNA的表達,以β-actin作為內參。每個待測基因設4個復孔。數據通過BIO-RAD CFX96系統進行處理,按照公式RQ=2-ΔΔCT計算各組間的倍數關系。
9. Western blot檢測NCOR、DNMT3b、IL-6和TNF-α mRNA的表達:收集細胞,加入300 μl蛋白裂解液,在冰上充分裂解,然后4 ℃、以離心半徑8 cm,12 000 r/min離心20 min。取上清液,測定蛋白濃度,并將所有樣本調整到相同濃度后,加入5×loading buffer,沸水中煮10 min使其變性。制備分離膠為8%的SDS-PAGE凝膠,每孔加樣40 μg進行電泳。300 mA濕轉150 min后,將PVDF膜室溫封閉1 h,應用NCOR(1︰2 000)、DNMT3b(1︰2 000)及GAPDH抗體(1︰10 000)的抗體4 ℃孵育過夜;再孵育放入辣根酶標記山羊抗兔的二抗(1︰50 000),用ECL化學發光法檢測NOCR和DNMT3b的表達水平。所得蛋白條帶圖片,使用Quality one軟件對圖像進行分析,以GAPDH的光密度值作為內參來校正各自目的蛋白的光密度值。

表1 基因引物序列
三、統計學方法
各實驗至少重復3次,使用SPSS 17.0對數據進行分析,兩組間進行t檢驗,多組之間進行單因素方差分析,P<0.05為具有統計學差異。
結 果
一、 LPS處理RAW264.7細胞后NCOR、IL-6和TNF-α表達以及NF-κB活性的變化
應用1 μg/ml LPS分別干預RAW264.7細胞0 h、24 h和48 h后,Western blot和real time-PCR檢測發現如圖1A和1B所示,LPS干預24 h和48 h后,RAW264.7細胞中NCOR蛋白和mRNA表達水平顯著低于0 h組(P<0.05),但24 h和48 h組NCOR蛋白和mRNA表達水平無顯著差異(P>0.05)。熒光素酶報告基因結果顯示如圖1C所示,LPS干預RAW264.7細胞后24 h、48 h,NF-κB 的活性較0 h組顯著升高(P<0.05),并且48 h組NF-κB 的活性顯著高于24 h(P<0.05)。此外,炎癥因子TNF-α和IL-6 mRNA的表達水平隨著LPS作用時間延長而升高(圖1D),呈時間依賴(P<0.05)。

圖1 LPS處理RAW264.7細胞后NCOR、IL-6和TNF-α表達以及NF-κB活性的變化,1 μg/ml LPS分別處理RAW264.7細胞0 h、24 h、48 h;注: A. Western blot檢測NCOR蛋白的表達水平;B: Real time-PCR檢測NCOR mRNA的表達水平;*與0 h比較,P<0.05;C:熒光素酶報告基因檢測NF-κB啟動子活性;*與0 h比較,P<0.05,^與24 h比較,P<0.05;D: Real time-PCR檢測IL-6和TNF-α mRNA的表達水平;*與0 h比較,P<0.05,^與24 h比較,P<0.05
二、LPS處理RAW264.7細胞后對NCOR基因啟動子甲基化的作用
應用MSP檢測發現,見圖2A: 1 μg/ml LPS干預RAW264.7細胞48 h后NCOR基因出現甲基化,提示LPS處理可誘導NCOR的啟動子甲基化。同時,利用Real time-PCR檢測NCOR mRNA的表達,見圖2B:發現LPS處理48 h后,NCOR mRNA的表達較對照組顯著降低(P<0.05)。但在使用DNA甲基化清除劑5′-aza和LPS同時處理后,NCOR mRNA的表達水平較對照組顯著降低(P<0.05),但較LPS組的表達水平有顯著的提升(P<0.05)。此外,還檢測了DNMT3b的蛋白水平(圖2C),發現其表達水平在LPS處理后較對照組顯著升高(P<0.05)。
三、下調DNMT3b對NCOR表達的影響
RAW264.7細胞分別轉染了3條siRNA和NC 48 h后,用Real time-PCR和Western blot檢測發現,3條DNMT3b siRNA均可以下調DNMT3b mRNA和蛋白的表達(P<0.05),其中DNMT3b siRNA-2的下調作用最為顯著(圖3A和3B)。因此,接下來的實驗均使用該條siRNA。進一步研究發現單獨轉染DNMT3b和NC,NCOR mRNA 和蛋白表達水平無顯著的差異(P>0.05)(圖3C和3D)。但RAW264.7細胞轉染DNMT3b siRNA 24 h后,再用LPS處理24 h后,NCOR的表達水平較NC+LPS組顯著降低,但仍然較單獨siRNA組顯著降低(P<0.05)。



圖2 LPS處理RAW264.7細胞對NCOR 啟動子甲基化的影響;注:A:MSP檢測1 μg/ml LPS處理細胞48 h后,NCOR 啟動子甲基化情況。U代表非甲基化NCOR DNA片段;M代表甲基化NCOR DNA片段;B:Real time-PCR檢測NCOR mRNA的表達水平。*與control組比較,P<0.05;^與LPS組比較,P<0.05;C: Western blot檢測1 μg/ml LPS處理細胞48 h后 DNMT3b蛋白的表達水平
四、下調DNMT3b對NF-κB活性及對IL-6和TNF-α表達的影響
利用熒光素酶報告基因檢測NF-κB啟動子活性, Real time-PCR檢測IL-6和TNF-α mRNA的表達水平,發現單獨DNMT3b siRNA組、NC+LPS組和DNMT3b siRNA +LPS組的NF-κB啟動子活性及IL-6、TNF-α mRNA的表達水平均顯著高于單獨NC組(P<0.05)。DNMT3b siRNA+LPS組的NF-κB啟動子活性及IL-6、TNF-α mRNA的表達水平較NC+LPS組明顯降低(P<0.05),但仍高于單獨DNMT3b siRNA組(P<0.05) (圖4A、B)。



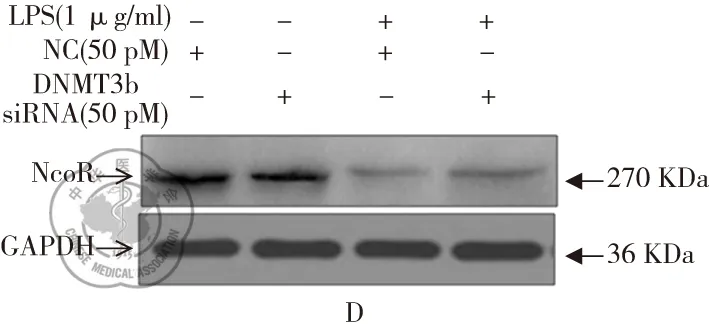
圖3 下調DNMT3b對NCOR表達的影響;注:A和B: Real time-PCR和 western blot檢測DNMT3b siRNA及NC轉染RAW264.7細胞后,NCOR mRNA和蛋白的表達水平,*與NC組比較,P<0.05;C和D: DNMT3b siRNA轉染24 h后再用1 μg/ml LPS處理24 h,Real time-PCR和 western blot檢測NCOR mRNA和蛋白的表達水平,*與NC組比較,P<0.05;^DNMT3b siRNA組比較,P<0.05;#NC+LPS組比較,P<0.05
討 論
本研究發現LPS可誘導巨噬細胞RAW264.7中NCOR的啟動子甲基化,進而下調NCOR的表達,提升NF-κB的活性,導致炎癥因子TNF-α、IL-6表達顯著上調。進一步研究表明DNMT3b參與了NCOR的啟動子甲基化。下調DNMT3b可部分逆轉LPS介導的NCOR甲基化,并抑制NF-κB的活性和炎癥因子TNF-α、IL-6表達的水平。上述研究說明,LPS介導NCOR的DNA 甲基化是促進巨噬細胞產生和釋放炎癥介質的關鍵環節。
失控性炎癥反應是ALI/ARDS發生、發展的重要環節,而巨噬細胞在其中發揮著重要的作用[17]。當內毒素刺激肺泡巨噬細胞時,LPS將激活細胞膜表面的Toll樣受體及下游信號通路NF-κB,產生大量的炎癥因子如TNF-α、IL-6和IL-1β以及ROS,進而導致組織損傷。本研究也發現了同樣的現象:在LPS刺激下,RAW264.7細胞的NF-κB的啟動子活性顯著增加以及炎癥因子TNF-α和IL-6表達上調。上述結果說明本研究LPS誘導巨噬細胞炎癥反應的模型是成功的,為進一步的研究奠定了基礎。


圖4 下調DNMT3b對NF-κB活性及IL-6和TNF-α表達的影響;注:A: 熒光素酶報告基因檢測NF-κB啟動子活性;B: Real time-PCR檢測IL-6和TNF-α mRNA的表達水平,*與NC組比較,P<0.05;^DNMT3b siRNA組比較,P<0.05;#NC+LPS組比較,P<0.05
NCOR作為核轉錄抑制因子在炎癥反應中發揮著重要作用。在正常狀態下,NCOR與一些炎癥通路的基因相結合,使它們處于抑制狀態[18]。當炎癥通路被激活后,NCoR被降解或從炎癥基因的啟動子上游離出來,消除對NF-κB、AP1等的轉錄抑制作用,進而釋放大量炎癥因子,加劇炎癥反應[17,19]。本研究中,LPS處理后,NCoR的表達顯著下降,而NF-κB的活性以及炎癥因子的表達水平顯著升高。而當NCOR的表達水平在DNMT3b siRNA處理下有所上調時,NF-κB的活性以及炎癥因子的表達水平同樣受到了部分抑制,這與文獻報道一致。上述研究說明LPS可調控NCOR的表達和定位,其可能的機制是通過激活泛素化途徑發揮作用[20-21]。有趣的是,本研究發現在LPS處理的巨噬細胞中NCOR發生了啟動子甲基化。當使用DNA甲基化清除劑5′-aza和LPS共同處理巨噬細胞時,NCOR的表達水平較之前的LPS單獨處理時有明顯的升高,再次說明LPS可誘導NCOR的啟動子甲基化,進而抑制其表達。
DNMTs是介導DNA甲基化的關鍵酶,其主要包括DNMT1、DNMT2和DNMT3三類。這三類酶由于mRNA剪切片段的不同又分為幾個亞型(如DNMT3a、3b)[22],不同的亞型發揮著不同的作用[23]。前期研究和其他研究說明在急性肺損傷模型中,DNMT3b在介導PPARγ等基因的啟動子甲基化中發揮著重要作用[15-16]。本研究同樣發現LPS可誘導DNMT3b的表達。而通過siRNA下調DNMT3b后,可部分顯著恢復下調的NCOR表達水平,進而逆轉了炎癥通路的激活和炎癥因子的上調。上述結果說明DNMT3b參與了LPS介導的NCOR 基因啟動子甲基化。
綜上所述,本研究進一步明確了NCOR在LPS誘導的巨噬細胞炎癥反應中的作用及機制,并首次發現了LPS介導NCOR基因的啟動子甲基化。研究結果為炎癥反應的發生、進展提供了新的理論基礎,為ALI/ARDS的防治提供了新的靶點和策略。
1 宋旸, 蔣昊翔, 張永紅, 等. 急性呼吸窘迫綜合征藥物研究進展[J/CD]. 中華肺部疾病雜志(電子版), 2015, 8(6): 769-772.
2 Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury[J]. N Engl J Med, 2005, 353(16): 1685-1693.
3 Zambon M, Vincent JL. Mortality rates for patients with acute lung injury/ARDS have decreased over time[J]. Chest, 2008, 133(5): 1120-1127.
4 Villar J, Sulemanji D, Kacmarek RM. The acute respiratory distress syndrome: incidence and mortality, has it changed?[J]. Curr Opin Crit Care, 2014, 20(1): 3-9.
5 Opal SM, Dellinger RP, Vincent JL, et al. The next generation of sepsis clinical trial designs: what is next after the demise of recombinant human activated protein C?*[J]. Crit Care Med, 2014, 42(7): 1714-1721.
6 Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome[J]. J Clin Invest, 2012, 122(8): 2731-2740.
7 Hong-Wei G, Lan L, De-Guo X, et al. NCoR negatively regulates adipogenic differentiation of mesenchymal stem cells[J]. In Vitro Cell Dev Biol Anim, 2015, 51(7): 749-758.
8 Wong MM, Guo C, Zhang J. Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation[J]. Am J Clin Exp Urol, 2014, 2(3): 169-187.
9 Heldring N, Nyman U, L?nnerberg P, et al. NCoR controls glioblastoma tumor cell characteristics[J]. Neuro Oncol, 2014, 16(2): 241-249.
10 Jennewein C, Kuhn AM, Schmidt MV, et al. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines[J]. J Immunol, 2008, 181(8): 5646-5652.
11 Barrès R, Yan J, Egan B, et al. Acute exercise remodels promoter methylation in human skeletal muscle[J]. Cell Metab, 2012, 15(3): 405-411.
12 Crevillén P, Yang H, Cui X, et al. Epigenetic reprogramming that prevents transgenerational inheritance of the vernalized state[J]. Nature, 2014, 515(7528): 587-590.
13 Qiu W, Baccarelli A, Carey VJ, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function[J]. Am J Respir Crit Care Med, 2012, 185(4): 373-381.
14 Chouliaras L, Mastroeni D, Delvaux E, et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer′s disease patients[J]. Neurobiol Aging, 2013, 34(9): 2091-2099.
15 Zhang XQ, Lv CJ, Liu XY, et al. Genome wide analysis of DNA methylation in rat lungs with lipopolysaccharide induced acute lung injury[J]. Mol Med Rep, 2013, 7(5): 1417-1424.
16 Lei C, Jiao Y, He B, et al. RIP140 down-regulation alleviates acute lung injury via the inhibition of LPS-induced PPARγ promoter methylation[J]. Pulm Pharmacol Ther, 2016, 37: 57-64.
17 Lin KL, Suzuki Y, Nakano H, et al. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality[J]. J Immunol, 2008, 180(4): 2562-2572.
18 Medzhitov R, Horng T. Transcriptional control of the inflammatory response[J]. Nat Rev Immunol, 2009, 9(10): 692-703.
19 Ogawa S, Lozach J, Jepsen K, et al. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation[J]. Proc Natl Acad Sci U S A, 2004, 101(40): 14461-14466.
20 Lu R, Hu X, Zhou J, et al. COPS5 amplification and overexpression confers tamoxifen-resistance in ERα-positive breast cancer by degradation of NCoR[J]. Nat Commun, 2016, 7: 12044.
21 Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells[J]. Nat Rev Immunol, 2010, 10(5): 365-376.
22 Xu F, Mao C, Ding Y, et al. Molecular and enzymatic profiles of mammalian DNA methyltransferases: structures and targets for drugs[J]. Curr Med Chem, 2010, 17(33): 4052-4071.
(本文編輯:黃紅稷)
郭小莉,劉霞,雷傳江,等. 脂多糖誘導巨噬細胞中核受體協同抑制因子啟動子甲基化對炎癥因子的調控作用[J/CD]. 中華肺部疾病雜志(電子版), 2017, 10(2): 157-162.
Lipopolysaccharide modulation the promoter methylation of nuclear receptor corepressor regulated inflammation mediators in macrophages
GuoXiaoli,LiuXia,LeiChuanjiang,WangGuansong,WangJianchun.
DepartmentofRespiratoryDisease,XinqiaoHospital,ThirdMilitaryMedicalUniversity,Chongqing400037,China
WangJianchun,Email:wjc5577@126.com
Objective To investigate the role and potential mechanism of nuclear receptor corepressor (NOCR) at lipopolysaccharide(LPS) mediation inflammation response in macrophages. Methods Western blot, realtime-PCR and luciferase assays was used to detect the expression of NCOR, interleukin-6(IL-6) and tumour necrosisfactor(TNF-α) and promoter activity of nuclear factor-κB (NF-κB) when RAW264.7 cells were treated with 1 μg/ml LPS. The promoter methylation of NCOR was analyzed by methylating-specific PCR(MSP) analysis, and the protein of DNMT3b was evaluated by western blot after cells stimulation with 1 μg/ml LPS for 48 h. Real time-PCR was also used to evaluate the expression of NCOR mRNA when RAW264.7 was treated with 1 μg/ml LPS combine with 1μM 5′-aza. After DNMT3b siRNA was transfected into RAW264.7 for 48 h, the mRNA and protein of DNMT3b was detected by western blot and real time-PCR. Additional, the expression of NCOR, TNF-α and IL-6 and NF-κB activity was analyzed after RAW264.7 was treated with 1 μg/ml LPS combine with DNMT3b siRNA. Results The expression of NCOR mRNA and protein was significantly decreased (P<0.05) and the expression of TNF-α and IL-6 mRNA, DNMT3b protein and activity of NF-κB was elevated (P<0.05) after cells was treated with 1 μg/ml LPS for 24 and 48 h, respectively. MSP assay showed LPS could modulate the NCOR promoter methylation. The expression of DNMT3b mRNA and protein was decreased after cells were transfected by DNMT3b siRNA. Additionally, down-regulation of DNMT3b was partly reversed LPS modulation the inhibition of NCOR, and decreased the expression of TNF-α and IL-6 mRNA, and activity of NF-κB (P<0.05). Conclusion NCOR promoter methylation is key step of occurrence and development of LPS mediation inflammation response. It might be a potential target for acute lung injury / acute respiratory distress syndrome (ALI/ARDS) treatment.
Nuclear receptor corepressor; DNA methylation; Nuclear factor-k-gene binding protein; Inflammatory factor
10.3877/cma.j.issn.1674-6902.2017.02.009
國家自然科學基金資助項目(81170066)
400037 重慶,第三軍醫大學新橋醫院呼吸內科
王建春,Email: wjc5577@126.com
R563
A
2017-02-07)
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年12期)2019-05-21 02:55:32
海峽科技與產業(2016年3期)2016-05-17 04:32:12
學苑創造·A版(2015年11期)2016-01-14 09:03:27
