GANT61對人胃癌細胞生長的影響及機制
2017-05-03 08:30:33米源杜媛鯤廖海江王林王雷
山東醫藥 2017年9期
關鍵詞:胃癌
米源,杜媛鯤,廖海江,王林,王雷
(1河北醫科大學第四醫院,石家莊050011;2河北醫科大學期刊社)
GANT61對人胃癌細胞生長的影響及機制
米源1,杜媛鯤2,廖海江1,王林1,王雷1
(1河北醫科大學第四醫院,石家莊050011;2河北醫科大學期刊社)
目的 探討鋅指蛋白(Gli)特異性抑制劑GANT61對人胃癌細胞AZ521和AGS生長的影響及機制。方法 用不同終濃度的GANT61處理AZ521和AGS 細胞72 h后,采用MTS法檢測GANT61對腫瘤細胞活力的影響;Western blotting法檢測GANT61作用于AZ521和AGS細胞24 h后Gli-1、Gli-2、CyclinD1和p21蛋白表達。流式細胞術檢測GANT61作用于AZ521和AGS細胞24 h后細胞周期。結果 GANT61作用于AZ521和AGS的IC50分別是7.94、12.70 μmol/L,GANT61作用后,AZ521和AGS細胞中Gli-1、Gli-2、CyclinD1蛋白表達顯著下調(P均<0.05),p21蛋白表達增加(P均<0.05)。GANT61處理AZ521和AGS細胞24 h后G0/G1期細胞所占比例增高(P均<0.05)。結論 GANT61可顯著抑制人胃癌細胞AZ521和AGS生長;其機制可能與下調Gli-1、Gli-2、CyclinD1蛋白表達,增加p21蛋白表達有關。
胃癌;鋅指蛋白特異性抑制劑;鋅指蛋白;細胞周期蛋白D1;p21蛋白;細胞周期
世界范圍內每年胃癌新發病例超過100萬,年死亡人數約達70萬[1]。研究發現,Hh通路的上下游出現異常活化均可致腫瘤的進展和轉移[2]。Hh通路在甲狀腺癌[3]、淋巴瘤[4]和惡性胸膜間皮瘤[5]等多種腫瘤組織中異常激活,與腫瘤的增殖和轉移密切相關。Yoo等[6]研究發現Hh信號通路在胃癌組織中過度活化且與胃癌發生發展及淋巴結轉移和預后相關,應用Hh通路抑制劑可下調胃癌細胞的侵襲能力。鋅指蛋白(Gli)1和Gli2作為Hh信號通路的下游關鍵核轉錄因子,起到轉錄激活作用,在多種腫瘤中異常高表達且參與腫瘤的發生發展。目前針對胃癌細胞中Hh下游核心靶點Gli1和Gli2與腫瘤生長轉移關系的研究較少。2016年3月,我們應用Gli的特異性抑制劑GANT61阻斷人胃癌細胞系AZ521和AGS細胞中Hh通路,觀察Gli對胃癌細胞生長作用的影響并初步探討GANT61抗腫瘤作用機制。
1 材料與方法
1.1 材料 人胃腺癌細胞系AZ521和AGS購自英國Sigma-Aldrich公司,伊格爾氏(EMEM)最低限度必需介質培養液、F12(Ham)液體培養液(美國Gibco公司),胎牛血清(美國Gemini公司),GANT61(美國Selleckchem公司),DMSO(美國MPBIO公司),CellTiter-Glo?發光法細胞活力檢測試劑盒(美國Promega公司),蛋白酶抑制劑(德國Roche公司),M-PER培養細胞總蛋白提取試劑、Pierce-BCA蛋白分析試劑盒(美國Thermo Scientific公司),Gli-1兔抗人多克隆抗體(美國Abcam公司),Gli-2兔抗人多克隆抗體、p21鼠抗人單克隆抗體(美國Thermo Fisher Scientific公司),細胞周期蛋白D1(CyclinD1)鼠抗人單克隆抗體(美國Cell signaling公司),GAPDH鼠抗人單克隆抗體(美國Santa Cruz公司),Mini-PROTEAN TGX 預制膠、Tris/甘氨酸/電泳緩沖液(美國BIO-RAD公司),PVDF膜(德國Millipore 公司),GloMax-96微孔板發光檢測儀(美國 Promega公司),Epics-XL 型流式細胞儀(美國Beckman-Coulter公司)。
1.2 細胞培養 將人胃癌細胞系AZ521培養于含10%胎牛血清和100 U/mL青-鏈霉素的EMEM最低限度必需介質培養基;AGS細胞培養于含10%胎牛血清和100 U/mL青-鏈霉素的F12(Ham)液體培養基,待細胞至70%~80%融合時傳代培養。
1.3 細胞活力測算 采用MTS法。將呈對數生長期的胃癌細胞,用0.25%胰蛋白酶消化后制成單細胞懸液,分別用含2%胎牛血清的EMEM重懸AZ521細胞和含2%胎牛血清的F12(Ham)液體培養液重懸AGS細胞,分別以50 μL培養基 /孔(5 000個細胞)接種于96孔培養板中,每個濃度設3個復孔;于37 ℃、5%CO2培養箱內培養過夜后,每孔加入50 μL不同終濃度的GANT61溶液(1.560 7、2.341 1、3.511 6、5.267 5、7.901 2、11.851 9、17.777 8、26.666 7、40 μmol/L),繼續培養72 h,并設空白對照DMSO組。72 h后取出96孔板室溫放置10 min,每孔加細胞活力檢測試劑100 μL,搖床輕搖30 s后置于避光處10 min,于GloMax-96微孔板發光檢測儀檢測,選擇CellTiter Glo軟件讀取數據后應用GraphpadPrism 6.1軟件分析,得出GANT61作用AZ521和AGS細胞72 h的IC50值。
1.4 AZ521和AGS細胞Gli-1 、Gli-2 、CyclinD1和p21蛋白表達檢測 采用Western blotting 法。六孔板內將AZ521(AZ521組)和AGS(AGS組)細胞更換含2%胎牛血清的培養基,加入終濃度為15 μmol/L的GANT61處理24 h,同時設置對照為DMSO組。24 h后用PBS洗滌細胞,每孔加細胞總蛋白提取試劑和蛋白酶抑制劑100 μL,用刮匙收取細胞的蛋白提取液,離心收集上清液,BCA法測定上清液總蛋白濃度。按照每樣本上樣量為10 μg總蛋白的標準于Mini-PROTEAN TGX 預制膠中上樣,在充滿Tris/甘氨酸/電泳緩沖液SDS的電泳槽中行電泳,實驗條件為200 V,60 min。電泳結束后在Tris-甘氨酸緩沖液中100 V,40 min后將蛋白電轉移印跡至PVDF膜上,TBST液洗膜3次,每次10 min,再用5%脫脂奶粉/TBST液封閉PVDF膜1 h后加一抗Gli-1濃度為1∶1 000,一抗CyclinD1濃度為1∶1 000,一抗p21濃度為1∶250,一抗GAPDH濃度為1∶10 000 ,4 ℃過夜,第2天TBST液洗膜3次,每次10 min后分別加抗兔和抗鼠二抗,二抗工作濃度均為1∶20 000,室溫孵育1 h后用TBST洗膜洗膜3次,每次10 min,ECL試劑置于PVDF膜上5 min發光,暗室行曝光、顯影。實驗重復3次。
1.5 細胞周期檢測 采用流式細胞術。于六孔板內加入終濃度為15 μmol/L的GANT61分別處理AZ521(AZ521組)和AGS(AGS組)細胞24 h,設置對照為DMSO組。48 h后用PBS洗滌細胞,胰酶消化,收集細胞制成單細胞懸液,70%乙醇固定6 h,調整樣品的細胞數為1×106/0.1 mL,加入碘化吡啶1 mL后在4 ℃冰箱染色30 min,上機測每組樣本的DNA含量。應用MuticycleAV分析軟件對每組樣本DNA細胞周期進行擬合分析,實驗重復3次。
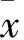
2 結果
2.1 GANT61對胃癌細胞活力的影響 GANT61作用AZ521和AGS細胞72 h后的IC50值分別是7.94、12.7 μmol/L。
2.2 兩組Gli-1、Gli-2、CyclinD1和p21蛋白表達比較 GANT61處理AZ521和AGS細胞24 h后發現Gli-1、Gli-2、CyclinD1蛋白表達顯著減少,p21蛋白表達顯著增加。AZ521組Gli-1、 Gli-2、CyclinD1蛋白表達分別為0.64±0.03、0.35±0.02、0.21±0.02、1.96±0.04,DMSO組分別為1.00±0.02、1.00±0.03、1.00±0.04、1.00±0.02,兩組Gli-1、Gli-2、CyclinD1、p21表達比較,P均<0.05。AGS組Gli-1、Gli-2、CyclinD1、p21蛋白表達分別為0.38±0.03、0.61±0.01、0.79±0.01、6.40±0.03,DMSO組分別為1.00±0.02、1.00±0.03、1.00±0.03、1.00±0.02,兩組4項指標比較,P均<0.05。
2.3 兩組細胞周期分布比較 AZ521組24 h后G0/G1期細胞比例為(71.33±4.51)%,與DMSO組(58±4)%比較顯著增高(P=0.019);AGS組24 h后G0/G1期細胞比例為(70.67±3.69)%,與DMSO組(52.67±3.79)%比較增高(P=0.004)。
3 討論
近年來研究發現胃癌中存在多條信號傳導通路異常激活的現象,其中針對Her-2等突變基因應用相應的靶向藥物取得較好效果[7],但隨其耐藥性的出現及部分患者存在Her-2陰性表達,迫使我們尋求新的有效治療靶點。Hh信號通路是一個高度協調且互相作用的網絡,主要包括Hedgehog配體、七次跨膜蛋白Smo、十二次跨膜受體Patched1 和Patched2、轉錄因子Gli家族(Gli1、Gli2、Gli3)等,通常Ptch與Smo形成復合物,抑制Smo活性,并抑制下游通路。其中Gli1和Gli2上調是Hh通路活化的最重要標志,而Gli3被認為是通路的轉錄抑制因子。除了經典的Hh通路活化途徑外,部分腫瘤亦存在非經典的不依賴Hh活化的Gli激活形式,對腫瘤細胞的周期調控和細胞增殖有重要意義[8],導致既往針對Hh通路的上游靶點Smo和Hh的抑制劑缺乏高效作用[9],而針對Gli靶點對Hh信號通路進行調控比針對Smo靶點有更好的抗腫瘤效果[10]。
細胞周期的調控與腫瘤發生密不可分,調控失常可致細胞獲得持續的增殖能力。G1~S及G2~M期是細胞周期的主要調控點。CyclinD1是調控G1至S期的最關鍵細胞周期蛋白,CyclinD1過表達可與CDK4結合形成復合物從而活化CDK4,促進腫瘤細胞G1到S期的轉換,促進腫瘤發生。細胞周期抑制蛋白p21是具CDK抑制活性的蛋白,其可與p27抑制CyclinD1活性從而導致細胞周期停止和阻斷細胞增殖。
本研究發現GANT61可顯著抑制胃癌AZ521和AGS細胞活力而起到抗胃癌作用。進一步研究發現,GANT61具有抗腫瘤細胞增殖的作用,GANT61組與DMSO組相比可阻滯更多腫瘤細胞于G0/G1期。GANT61分別作用AG521和AGS后CyclinD1表達明顯降低,p21表達明顯升高,推測GANT61調控細胞周期和抗腫瘤機制可能是Hh/Gli信號通路在胃癌細胞中處于活化狀態并參與了腫瘤細胞的增殖過程,GANT61可通過特異性抑制胃癌細胞的Gli-1和Gli-2表達從而下調下游靶基因CyclinD1蛋白表達和升高p21蛋白表達來實現對AZ521和AGS細胞的生長抑制作用。
綜上所述,GANT61有顯著抗腫瘤作用,其抗胃癌作用機制可能與其下調Gli1和Gli2蛋白表達從而抑制腫瘤細胞的活力及阻滯細胞周期等有關。
[1] Jemal A, Bray F, Center MM, et al. Global cancer statistics[J]. CA Cancer J Clin, 2011,61(2):69-90.
[2] Ng JM, Curran T. The Hedgehog's tale: developing strategies for targeting cancer[J]. Nat Rev Cancer, 2011,11(7):493-501.
[3] Dong W, Cui J, Tian X, et al. Aberrant sonic hedgehog signaling pathway and STAT3 activation in papillary thyroid cancer[J]. Int J Clin Exp Med, 2014,7(7):1786-1793.
[4] Kern D, Regl G, Hofbauer SW, et al. Hedgehog/GLI and PI3K signaling in the initiation and maintenance of chronic lymphocytic leukemia[J]. Oncogene, 2015,34(42):5341-5351.
[5] Felley-Bosco E, Opitz I, Meerang M. Hedgehog Signaling in Malignant Pleural Mesothelioma[J]. Genes (Basel), 2015,6(3):500-511.
[6] Yoo YA, Kang MH, Lee HJ, et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer[J]. Cancer Res, 2011,71(22):7061-7070.
[7] Fu YF, Gui R, Liu J. HER-2-induced PI3K signaling pathway was involved in the pathogenesis of gastric cancer[J]. Cancer Gene Ther, 2015,22(3):145-153.
[8] Gurung B, Feng Z, Hua X. Menin directly represses Gli1 expression independent of canonical Hedgehog signaling[J]. Mol Cancer Res, 2013,11(10):1215-1222.
[9] Dlugosz A, Agrawal S, Kirkpatrick P. Vismodegib[J]. Nat Rev Drug Discov, 2012, 11(6):437-438.
[10] Li H, Lui N, Cheng T, et al. Gli as a novel therapeutic target in malignant pleural mesothelioma[J]. PLoS One, 2013,8(3):e57346.
河北省醫學科學研究重點課題(20160177)。
王雷(E-mail:yuankundu@163.com)
10.3969/j.issn.1002-266X.2017.09.010
R735.2
A
1002-266X(2017)09-0034-03
2016-08-10)
猜你喜歡
昆明醫科大學學報(2022年1期)2022-02-28 07:43:36
昆明醫科大學學報(2021年5期)2021-07-22 07:32:22
基層中醫藥(2020年2期)2020-07-27 02:46:06
中國組織化學與細胞化學雜志(2016年3期)2016-02-27 11:15:35
中國衛生標準管理(2015年3期)2016-01-14 03:41:46
中外醫療(2015年18期)2016-01-04 06:51:55
吉林大學學報(醫學版)(2015年4期)2015-12-17 07:48:25
醫學研究雜志(2015年6期)2015-07-01 17:40:49
醫學研究雜志(2015年9期)2015-07-01 17:28:27
中國當代醫藥(2015年20期)2015-03-01 02:04:29
