氣相色譜法檢測平菇中9種農藥殘留分析
2017-04-28 03:28:27王潔蓮
農學學報 2017年3期
王潔蓮,牛 瑋,孫 靜
(1山西省農產品質量安全檢驗監測中心,太原030025;2山西省運城市鹽湖區種子管理站,山西運城044000)
氣相色譜法檢測平菇中9種農藥殘留分析
王潔蓮1,牛 瑋1,孫 靜2
(1山西省農產品質量安全檢驗監測中心,太原030025;2山西省運城市鹽湖區種子管理站,山西運城044000)
為解決食用菌中的農藥殘留檢測問題,以基地生產的平菇樣品為實驗材料,采用固相萃取方法對平菇中白菌清、三唑酮等9種農藥殘留的檢測方法進行研究,用石墨碳黑氨基柱凈化樣品,利用氣相色譜作為檢測手段,ECD檢測器進行定性定量分析。研究表明,平菇中9種農藥的含量重復性實驗相對標準偏差為2.05%~6.35%,檢出限為0.0003~0.005 mg/kg,定量限為0.0009~0.02 mg/kg。9種農藥在0.05~2.0 mg/L范圍內線性關系良好,r值均大于0.990。此方法檢測平菇中9種農藥殘留分析準確、可行。
農藥殘留;平菇;氣相色譜法
0 引言
平菇的醇香備受人們的鐘愛,是餐桌上的常見食用菌。食用菌的生產環境不同于蔬菜,根據GB 2763—2014規定[1],食用菌與蔬菜不同類,食用菌一般是用適宜的菌種在適宜的環境下人工栽培而成。在平菇的生長過程中,會因環境的改變產生很多雜菌和蟲害,所以必須使用農藥。不按規定使用農藥,或使用農藥后在間隔期上市是平菇中農藥殘留超標的主要問題。目前常用的平菇的檢測方法有質譜法和氣相色譜法。質譜儀器價格貴,運行成本高,對操作人員要求嚴格,不適于檢測參數的大面積普及。目前的氣相色譜法檢測參數分散,根據無公害檢測目錄規定,平菇檢測的有機氯和菊酯類農藥共有5種,需使用3種檢測方法[2-4],這3種檢測方法所使用的前處理技術有液液萃取和填充柱法,每種檢測方法的前處理過程均不相同,檢測參數少,耗時長,對環境污染嚴重,不能為農產品質量安全提供簡便快捷的手段。筆者用乙腈提取,氣相色譜法(GC-ECD)檢測,比較3種固相萃取柱和吸附劑凈化區別,選擇出穩定、可靠、靈敏度高、滿足要求的平菇中9種農藥殘留同時檢測的氣相色譜方法,為平菇檢測參數的普及提供必要的監測手段。
1 材料與方法
1.1 前處理材料
1.1.1 樣品來源 呂梁示范基地栽培的平菇樣品。
1.1.2 試劑和標準品 氯化鈉(天津北辰方正試劑廠)于650℃灼燒2 h后備用,乙腈、正已烷、甲苯、丙酮均為色譜純(迪馬科技);石墨炭黑氨基固相萃取柱(1000 mg/mL)、氨基固相萃取柱(500 mg/mL)、氟羅里矽柱(500 mg/mL),均為美國Agilent公司。標準品為百菌清、三唑酮、腐霉利、聯苯菊酯、甲氰菊酯、氯氟氰菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯,濃度為100 mg/L,均購自農業部環境監測總站(天津)。
1.1.3 主要儀器 Agilent7890B氣相色譜儀(美國安捷倫公司),μ-ECD檢測器;TP-2101電子天平(丹佛儀器有限公司);T25basic高速勻漿機,RV05basic旋轉蒸發儀,TKA MS3旋窩混勻器(德國IKA公司);TTL-DCⅡ型氮吹儀(北京同泰公司)。
1.2 檢測方法
1.2.1 樣品的提取與凈化 準確稱取20.00 g平菇樣品于200 mL玻璃燒杯中,準確加入40.0 mL乙腈,高速勻漿2 min(10000 r/min)后抽濾,用含5~7 g氯化鈉(已烘)的100 mL具塞量筒收集濾液30~40 mL,加塞密閉,充分振蕩1 min,去塞,在室溫下靜置30 min以上;吸取10 mL上層乙腈相于150 mL雞心瓶中,于40℃下旋蒸濃縮至近干,加乙腈:甲苯(3:1)5 mL待凈化。
凈化:用6.0 mL乙腈:甲苯(3:1)混合溶劑預淋洗石墨炭黑氨基柱,當溶液接近柱表面時,加入上述凈化液,再分4次用5 mL乙腈:甲苯(3:1)混合溶劑洗瓶,收集洗脫液用于45℃旋蒸濃縮至1 mL左右,用氮氣吹干,5 mL正已烷準確定容,待測。
1.2.2 氣相色譜條件 中級性毛細管柱DB-17(30 m× 0.25 mm×0.25 mm);程序升溫:初溫150℃,保持1 min,以10℃/min升到270℃,保持15 min,以20℃/min升到280℃,保持2 min;柱流量2 mL/min。檢測器μ-ECD,溫度300℃;進樣口溫度235℃;進樣模式分流,進樣量1μL,分流比10:1;尾吹流量60 mL/min。
1.2.3 標準溶液配制 準確移取1.00 mL的百菌清、三唑酮、腐霉利、聯苯菊酯、甲氰菊酯、氯氟氰菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯單個農藥標準溶液,用正已烷做溶劑,配制成10 mg/L的農藥混合標準儲備液(貯存在-16℃以下的冰箱中)。上機之前,用正已烷將10 mg/L混合工作儲備液依次稀釋為2.0、1.0、0.5、0.2、0.1、0.05 mg/L的混合工作使用液。
1.3 標準曲線的繪制
將配制好的0.05、0.1等一系列的6個濃度點的工作使用液依據1.2.2章節的氣相色譜條件,以質量濃度(x)為橫坐標,峰面積(y)為縱坐標繪制標準曲線,每個濃度5次重復。
1.4 準確度和精密度的測定
在陰性平菇樣品中分別添加0.05、0.1、1.0 mg/kg的3個水平的9種農藥的混合工作溶液,每個水平重復5次,按1.2.2章節的氣相色譜條件測定回收率及相對標準偏差(RSD),見表1。
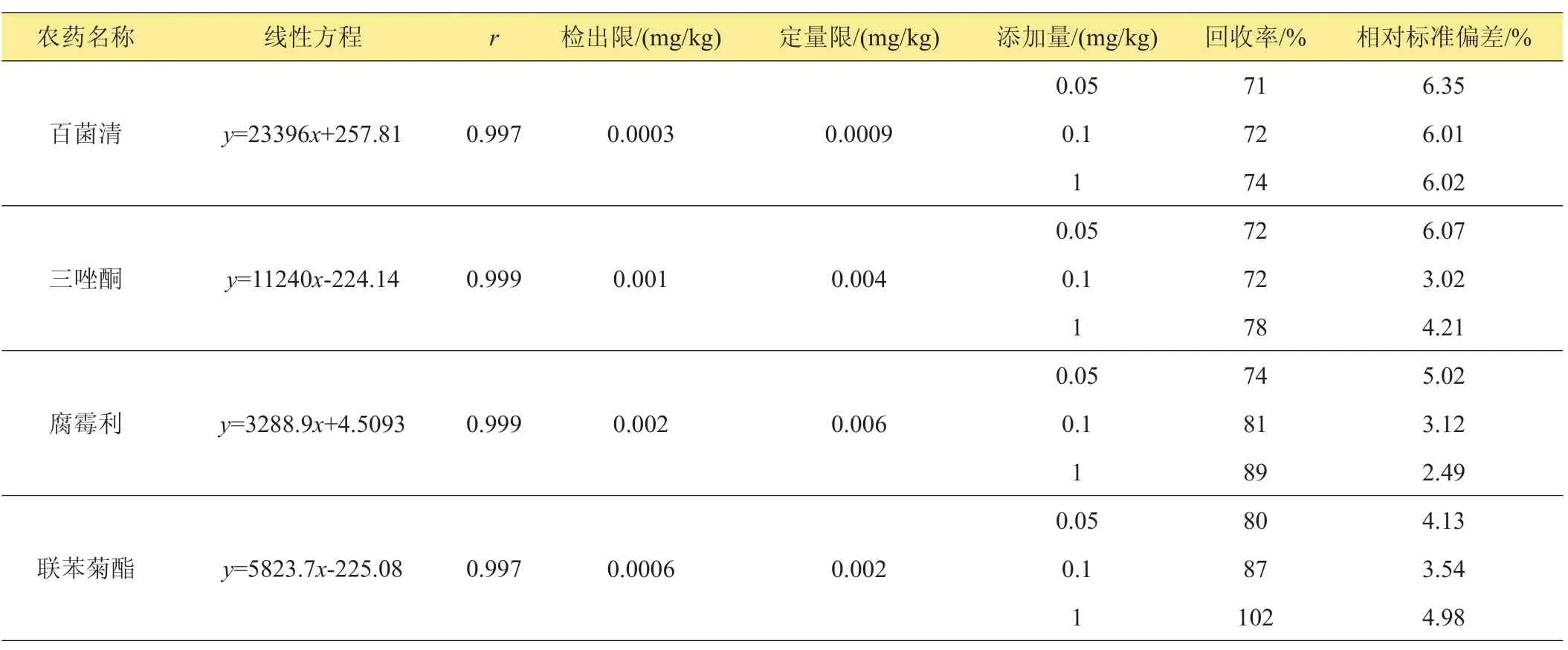
表1 9種農藥的線性方程、相關系數、檢出限、定量限、回收率及相對標準偏差(n=5)
2 結果與分析
2.1 提取溶劑的選擇
依據極性的大小,本著相似相溶的原則,查閱參考文獻[5-7],選擇丙酮、甲醇、乙腈對樣品進行提取。丙酮、甲醇提取樣品時提取液顏色深,影響色譜柱的使用壽命,對色譜系統污染嚴重。乙腈提取樣品,雜質少,顏色淺,回收率滿意。
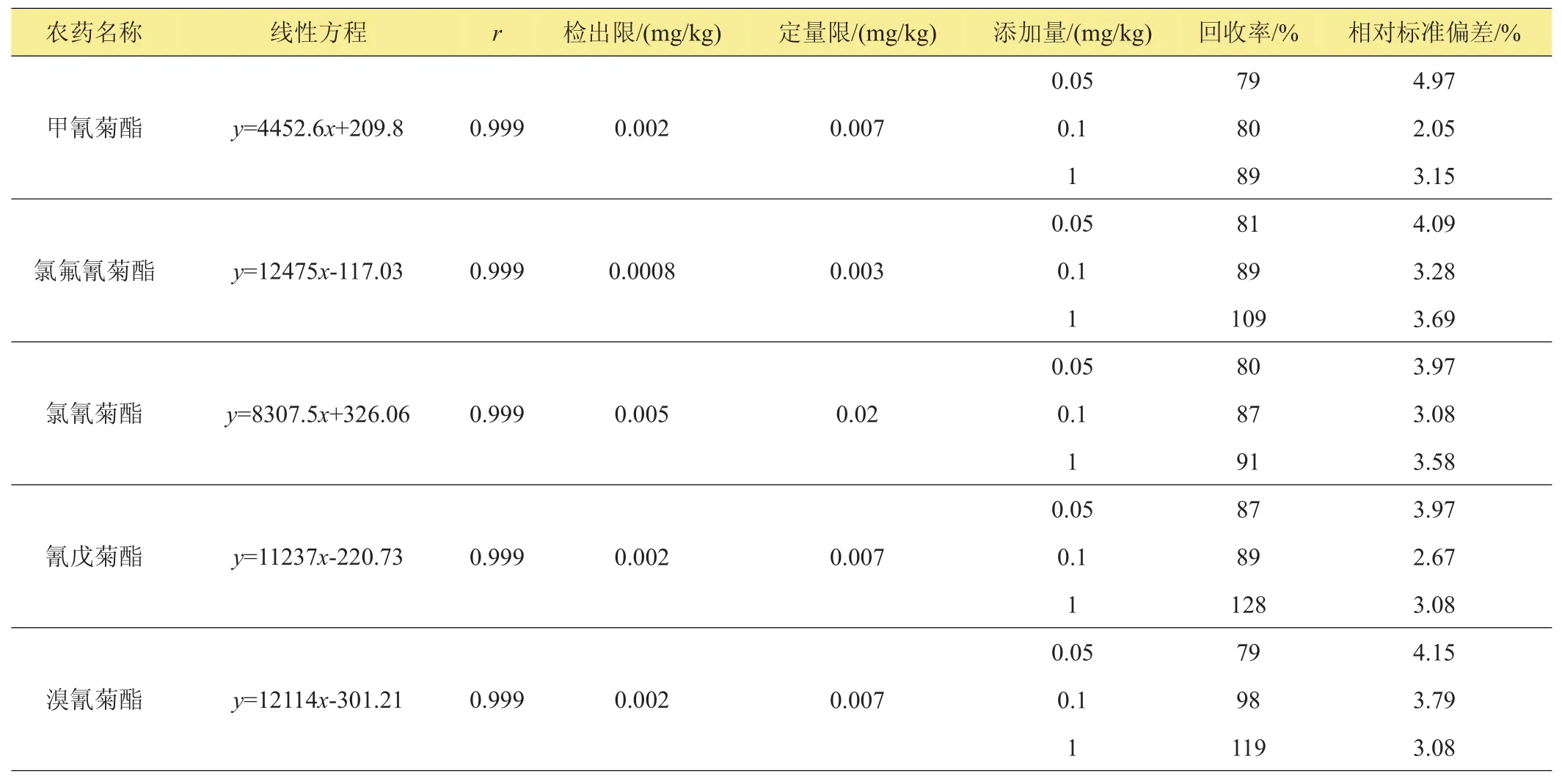
續表1
2.2 凈化條件的選擇
現行的凈化方法是吸附劑(PSA/C18/GCB)凈化[8-13]和SPE柱凈化[14-19],吸附劑凈化應用于平菇的氣相色譜檢測,不能有效減少干擾色譜峰。SPE柱凈化是目前氣相色譜檢測中最常用的凈化方法。SPE柱中的氨基柱、氟羅里矽柱和石墨炭黑氨基柱在這幾種農藥檢測中有使用的報道。查閱現有的檢測標準[1-7]和文獻[20-22],進行單因素水平實驗,確定各凈化柱的洗脫溶液和體積,設計3因素3水平的正交實驗見表2,以9種農藥的平均回收率為參考指標,結果見表3。可以看出A1B3C3、A2B1C2和A3B2C13種組合的回收率水平在70%~130%,為了確保每個農藥的回收率都符合要求,根據回收率水平70%~130%、最接進100%回收率的要求,確定出最佳凈化條件A2B1C2,按照這個條件進行樣品前處理,9種農藥得到良好的分離,在28 min內完成檢測。空白樣品見圖1。
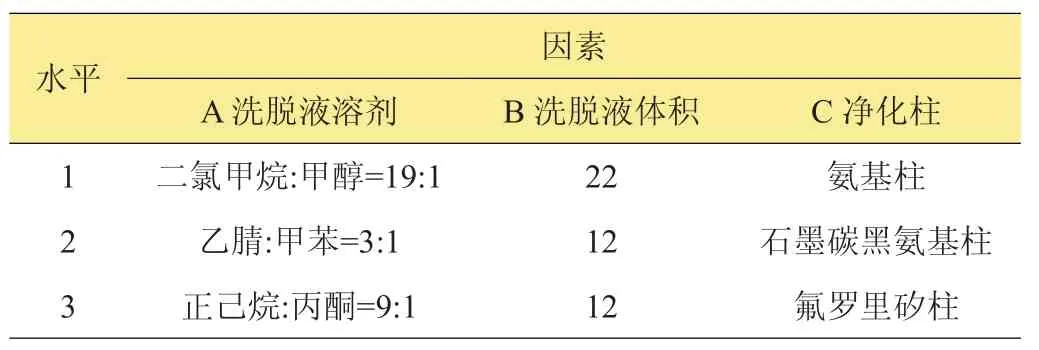
表2 9種農藥殘留正交實驗因素水平表

表3 9種農藥殘留正交因素實驗結果表
2.3 線性方程、檢出限和定量限
用正己烷溶劑作為9種農藥的標準品稀釋液,依次稀釋為2.0、1.0、0.5、0.2、0.1、0.05 mg/L一系列6個濃度點的混合標準溶液,用氣相色譜ECD進行檢測,外標法定量。9種農藥的6個濃度與峰面積有良好的線性關系。在最低添加濃度0.05 mg/kg下,以相同的時間段計算,3倍的信躁比為檢出限,10倍的信躁比為定量限。9種農藥的線性方程、檢出限及定量限見表1。
圖1 空白樣品色譜圖
2.4 方法的準確度和精密度
結果表明,在0.05~1.0 mg/kg 3個添加水平下,平均回收率為71%~119%,相對標準偏差為2.05%~6.35%,定量限為0.0009~0.02 mg/kg,檢出限為0.0003~0.005 mg/kg,說明本方法靈敏度高,穩定性好,可作為平菇中9種農藥殘留的檢測方法。
在已知的陰性平菇樣品中分別加入9種農藥的混合標準儲備液,每個加標樣品做3個平行實驗,重復測定5次,計算平均回收率為70%~119%,相對標準偏差不大于7%(表1),說明本方法準確可靠。圖2為標準溶液色譜圖,圖3為樣品添加色譜圖。
2.5 實際樣品分析
按照本方法比較后選定的前處理方法和儀器條件對呂梁示范基地的平菇樣品在上市前進行檢測,共檢測樣品50個。隨機抽取20個樣品做加標回收率實驗,加標濃度為0.1、1.0 mg/kg,回收率為71%~119%,說明本方法能滿足實際樣品的分析要求。

圖2 標準溶液色譜圖

圖3 平菇加標樣品色譜圖
3 結論
筆者先比較了吸附劑和固相萃取柱兩種凈化方式,確定固相萃取柱凈化,然后再設計3因素3水平的正交實驗,比較不同的溶劑、溶劑的用量在石墨炭黑氨基柱、氨基柱和氟羅里矽柱3種固相萃取柱凈化平菇樣品的區別,最終確定了白菌清等9種農藥在平菇中的氣相色譜儀上的檢測方法。本方法選用石墨炭黑氨基柱對平菇樣品進行凈化,該方法準確、靈敏度高,滿足國家要求;本方法為目前普及的氣相色譜提供了檢測參考,為平菇的質量安全增加了檢測方法;本方法補充了平菇樣品中的農藥多殘留檢測方法,有望成為地方標準。
4 討論
食用菌作為單列類(與蔬菜不同類),目前農藥多殘留檢測的方法是質譜法[7-9],氣相色譜方法涉及的多殘留檢測使用的前處理方法不統一,一個參數一個方法。筆者將食用菌常檢的農藥用同一種前處理方法凈化,以平菇為樣品,一次處理可同時檢測9個參數,平菇中農藥殘留氣相色譜檢測的報道文獻[22-25]最多檢測5種農藥。本研究的不足之處是石墨炭黑氨基柱凈化使用的淋洗液用量大,增加了實驗時間,有待進一步研究。
[1]中華人民共和國國家衛生和計劃生育委員會,中華人民共和國農業部.GB 2763—2014,食品安全國家標準食品中農藥最大殘留限量[S].北京:中國標準出版社,2014:11-25.
[2]中國疾病預防控制中心營養與食品安全所,北京是疾病預防控制中心,等.GB/T 5009.146—2008,植物性食品中有機磷和擬除蟲菊酯類農藥多種殘留量的測定[S].北京:中國質檢出版社,2008:3-4
[3]農業部環境質量監督檢驗測試中心(天津),農業部環境保護科研監測所.NY/T 761—2008,蔬菜和水果中有機磷、有機氯、擬除蟲菊酯和氨基甲酸酯類農藥多殘留的測定[S].北京:中國標準出版社,2008:16-28.
[4]中華人民共和國秦皇島出入境檢驗檢疫局.GB/T 19648—2006,水果和蔬菜中500種農藥及相關化學品殘留量的測定氣相色譜-質譜法[S].北京:中國質檢出版社,2006:25.
[5]張太成.食用菌中農藥多殘留研究[D].長春:吉林農業大學,2007.
[6]常巧英.食用菌中農藥多殘留LC-MS/MS測定方法研究[D].泰安:山東農業大學,2014.
[7]中華人民共和國秦皇島出入境檢驗檢疫局,山東農業大學,等.GB/ T 23216—2008,食用菌中503種農藥及相關化學品殘留量的測定氣相色譜-質譜法[S].北京:中國質檢出版社,2008:21.
[8]中華人民共和國秦皇島出入境檢驗檢疫局,山東農業大學.GB/T 23202—2008,食用菌中440種農藥及相關化學品殘留量的測定液相色譜-串聯質譜法[S].北京:中國質檢出版社,2008:10
[9]中華人民共和國秦皇島出入境檢驗檢疫局,山東農業大學.GB/T 20769—2008,水果和蔬菜中450種農藥及相關化學品殘留量的測定液相色譜-串聯質譜法[S]北京:中國標準出版社,2008:3
[10]肖海軍,張偎,陳孝權,等.分散固相萃取-氣質聯用法同時檢測茶葉中32種農藥多殘留[J].西南師范大學學報:自然科學版,2015,40 (12):28-32.
[11]褚能明,楊俊英,柴勇,等.兩種SPE凈化方法在蔬菜農藥多殘留分析中的比較[J].西北農業學報,2013,23(5):1525-1528.
[12]王潔蓮,董燕飛.高效液相色譜法同時檢測蔬菜中的7種農藥殘留的分析[J].中國農學通報,2015,31(12):267-272.
[13]王潔蓮,閻會平.氣相色譜法同時檢測蘋果中的6種農藥殘留的分析[J].食品研究與開發,2015,36(19):155-157.
[14]王潔蓮,閻會平.氣相色譜法檢測橙子中氟蟲腈農藥殘留分析[J].農學學報,2016,6(1):83-87.
[15]徐虹,曹文婷,易承學,等.液液萃取氣相色譜法測定飲用水中百菌清等12種農藥殘留[J].中國衛生檢驗雜志,2015,24(23):3363-3365.
[16]Chen X S,Bian Z Y,Yang F,et al.Comparison of three different QuEChERS sample treatment methods in the analysis of more than one hundred pesticide residues in tobacco by gas chromatographytandem mass spectrometry[J].Chinese Journal of Chromatography, 2013,11(31):1116-1128.
[17]王潔蓮,牛瑋,劉巍,等.蔬菜中農藥殘留檢測標準及其檢測方法概述[J].食品安全質量檢測學報,2016,7(4):1587-1592.
[18]王天嬌,吳平谷,胡爭艷,等.固相萃取-超高效液相色譜法檢測保健食品中維生素D3[J].中國衛生檢驗雜志,2016,7(14):128-131.
[19]閆震,聶繼云,徐國鋒,等.超高效液相色譜-串聯質譜法對比4種凈化方式對不同色素含量基質中19種農藥殘留檢測的影響[J].分悉測試學報,2014,33(9):1000-1009.
[20]王潔蓮,田子卿.液相色譜法檢測韭菜中阿維菌素殘留方法的改進[J].食品安全質量檢測學報,2015,6(1):289-292.
[21]尚天翠.超聲波法提取曼陀羅種子總生物堿的工藝優化[J].遼寧化工,2016,30(5):537-539.
[22]柳佳,陳國寶,宋桂萍,等.正交試驗法優選降脂合劑的制備工藝[J].現代中藥研究與實踐,2016,21(4):31-34.
[23]王潔蓮,牛瑋,劉巍.氣相色譜法測定圓茄子中氟啶脲[J].食品安全質量檢測學報,2014,5(7):2215-2219.
[24]劉永明,葛娜,崔宗巖.糧谷中農藥殘留前處理技術的研究進展[J].食品安全質量檢測學報,2014,5(7):2151-2160.
[25]周長紅.氣相色譜法測定平菇中16種有機磷農藥殘留量[J].中國食用菌,2013,32(1):35-37.
Determination of 9 Pesticide Residues in Oyster Mushroom by Gas Chromatography
Wang Jielian1,Niu Wei1,Sun Jing2
(1Agricultural Products Quality Safety Monitoring Center of Shanxi Province,Taiyuan 030025,Shanxi,China;2Salt Lake Valley Seed Station of Yuncheng City,Yuncheng 044000,Shanxi,China)
To detect pesticide residues in edible mushrooms,oyster mushroom samples were taken as the experiment material.The solid-phase extraction method was adopted to study the detection methods for 9 pesticide residues such as chlorothalonil and triadimefon in oyster mushrooms.The samples were extracted with acetonitrile,further cleaned-up by graphite carbon amino columns and detected by gas chromatography with electron capture detector(GC-ECD).The result showed that the relative standard deviations of nine pesticides content were 2.05%-6.35%under this method,the limits of detection were 0.0003-0.005 mg/kg and the limits of quantification were 0.0009-0.02 mg/kg.The nine pesticides had a good linearity from 0.05 to 2.0 mg/L and the r values were greater than 0.990.The method is feasible and accurate for determining nine residues in oyster mushroom.
Pesticide Residues;Oyster Mushroom;Gas Chromatography
S657.71,TQ450.26
:A論文編號:cjas16080024
山西省科技攻關項目“農藥殘留與農田鼠害防治技術研究”(20130310011-4)。
王潔蓮,女,1980年出生,山西臨猗人,高級農藝師,碩士,研究方向:農產品中農藥殘留的檢測。通信地址:030025山西省太原市晉源新區景明北路5號山西省農產品質量安全檢驗監測中心,E-mail:agilent2006@163.com。
2016-08-29,
:2016-12-02。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
