硫代砷化物的合成、鑒定和定量分析方法研究
2017-04-25 02:58:40王敏黛郭清海
分析化學 2016年11期
王敏黛+郭清海


摘要 制備了4種硫代砷酸鹽,采用電噴霧高分辨質譜(ESIHRMS)進行鑒定,確證為一硫代砷酸鹽(HnAsSOn-33)、二硫代砷酸鹽(HnAsS2On-32)、三硫代砷酸鹽(HnAsS3On-3)和四硫代砷酸鹽(HnAsS4n-3)。以合成的硫代砷酸鹽為標準品,建立了基于離子色譜電感耦合等離子質譜(ICICPMS)同時測定4種硫代砷酸鹽、亞砷酸鹽和砷酸鹽的方法。本方法線性范圍為3.3~833μg/L,線性相關系數R2>0.99,方法檢出限為0.4~25μg/L。本研究合成的硫代砷酸鹽標準物質及建立的定量分析方法,對后續富含硫化物的天然水體中不同形態砷的遷移和轉化研究具有重要意義。
關鍵詞硫代砷化物;合成;鑒定;電噴霧高分辨質譜;離子色譜電感耦合等離子體質譜;定量分析
1引言
作為危害性最大的致癌物質之一,環境介質(水、土壤、農作物等)中砷經富集并被人體長期攝入可導致不同癥狀的慢性砷中毒。天然高含量砷的水的廣泛存在是環境砷異常的重要表現之一,已使全球數億人受到威脅。目前,天然水中砷的地球化學和環境歸宿研究通常將砷酸鹽、亞砷酸鹽、甲基砷和二甲基砷等作為砷的主要形態。然而,近年來對富硫化物天然水的研究發現,具備巖漿熱源的高溫水熱系統在巖漿脫氣過程的影響下,所排泄的地熱水(熱泉或地熱井流體)常同時富集硫化物和砷,硫代砷化物已被證明是冰島Geysir等地熱區的高溫地熱水中砷的主要存在形式,此類富砷/富硫地熱水中的砷有相當比例以硫代砷化物的形式存在[1~3\]。在以水熱形式存在的地熱資源的開發利用過程中,地熱流體中高含量砷對地表水的污染[4\]也受到了研究者的密切關注,但硫代砷化物作為流體中砷的主要形態,卻長期被研究者忽視。硫代砷化物種類繁多,包括多種硫代砷酸鹽和硫代亞砷酸鹽,以及不同類型的甲基硫代砷化物。與砷的常見形態不同,各類硫代砷化物的毒性和生物毒理作用大不相同[5,6\]。PlanerFriedrich等[6\]在進行硫代砷化物對費氏弧菌的毒理效應研究中發現,硫代砷化物的毒性與其中SH官能團的數量呈正相關;一硫代砷化物和二硫代砷化物的毒性遠小于三硫代砷化物,而三硫代砷化物的毒性則與亞砷酸鹽相當。曹煊等[7\]發現人體肝細胞在砷代謝的過程中可產生一甲基硫代砷酸,而人體代謝砷的過程可認為是砷的毒性降低的過程,這顯示甲基硫代砷化物的毒性相對較低。硫代亞砷酸鹽僅在極其嚴苛的條件下才能穩定存在。即便在厭氧條件下,如水環境偏堿性(水中SH
含量小于OH
),硫代亞砷酸鹽也不能長時間穩定存在,會轉化為亞砷酸鹽;而在有氧條件下,硫代亞砷酸鹽則迅速轉化為硫代砷酸鹽[8\]。綜上所述,硫代亞砷酸鹽主要存在于強還原/高硫化物/非強堿性地下水環境中,一旦地下水升流至近地表偏氧化環境或泉口,即轉化為硫代砷酸鹽。因此,針對天然水中的砷在地表的環境地球化學過程研究,無需考慮硫代亞砷酸鹽形態。
天然水樣品中硫代砷化物檢測技術是開展其環境地球化學研究的必要前提。Schwedt等[9\]通過毛細管電泳和離子色譜分離檢測不同形態的硫代砷化物,分離時間長達5h。其后,加拿大、德國和冰島的研究團隊優化了測試方法[10~12\],采用離子色譜分離不同形態硫代砷化物并利用原子熒光在線檢測,但該技術僅用于內部研究,尚未向外界開放。國內尚未見關于天然水環境中硫代砷化物研究的文獻報道。因此,建立水體中不同形態硫代砷化物的同時檢測方法具有重要意義。此外,硫代砷化物的存在也對砷的環境地球化學行為有重要影響,很多科學問題亟待解決。例如,熱泉排泄后其中硫代砷化物將進一步在富氧大氣環境下發生形態轉化,氧化還原條件的變化、含硫/鐵礦物的沉淀、光催化效應等將對砷的不同存在形態的穩定性產生重要影響,而當前缺乏針對上述過程影響下熱泉環境中硫代砷化物形態轉化的系統研究。目前,已開展了一些針對天然水系統中砷、硫、鐵地球化學過程的研究[13~18\],但對于熱泉環境中硫代砷化物在砷、硫、鐵地球化學耦合過程(控制砷的環境地球化學遷移的主要過程)中的作用卻所知甚少。
本研究建立了水中不同形態硫代砷酸鹽的同時定量分析方法。目前,尚無商品化的硫代砷酸鹽,因此本研究通過改進一硫代砷酸鹽、二硫代砷酸鹽、四硫代砷酸鹽[11\]和三硫代砷酸鹽[19\]合成方法,合成了高純度的4種硫代砷酸鹽,并采用電噴霧高分辨質譜進行了確證。以合成的4種硫代砷酸鹽以及商品化的砷酸鹽和亞砷酸鹽為標準樣品,建立了基于離子色譜電感耦合等離子體質譜同時定量測定4種硫代砷化物及砷酸鹽和亞砷酸鹽的方法。
2實驗部分
2.1儀器與試劑
ICS900型離子色譜儀(美國ThermoFisher公司);7700x型電感耦合等離子體質譜儀(ICPMS,美國Agilent公司);DionexIonPacAS19陰離子交換柱(250mm×4mm,10μm)和保護柱AG19(50mm×4mm,10μm)(美國ThermoFisher公司);KQ5200DE型數控超聲波清洗器(昆山市超聲儀器有限公司)。
升華硫(化學純,天津市科密歐化學試劑有限公司);Na3AsO3(分析純,西亞試劑);Na2S、As2O3(分析純,上海統亞化工科技發展有限公司);NaOH(分析純,國藥集團化學試劑有限公司);As2S5(優級純,JP德國SigmaAldrich公司);As(III)(GBW08666,純度≥98.5%,中國計量科學研究院);As(V)(GBW08667,純度≥98.1%,中國計量科學研究院)。
2.2硫代砷化物制備
一硫代砷酸鹽:將5.00gAs2O3和6.00gNaOH溶于20mL脫氣水,加入1.44g單質硫,在回流裝置中加熱至100℃,保持2h。過濾溶液中多余的單質硫,濾液在4℃下冷卻,真空干燥。將晶體溶解于脫氣水中,加乙醇使其重結晶,過濾、干燥得到一硫代砷酸鈉(晶體化學結構式:Na3AsSO3·7H2O,溶液中存在形式HnAsSOn-33)。
二硫代砷酸鹽:上述步驟中單質硫的加入量更換為5.76g,可得二硫代砷酸鈉晶體(晶體化學結構式:Na3AsO2S2·7H2O;溶液中存在形式:HnAsS2O2n-3)。
三硫代砷酸鹽:將0.2gNa3AsS4·8H2O溶于20mL0.1mol/LNaOH,完全溶解后,加入0.244gBaCl2晶體(BaCl2·8H2O),過濾,濾液真空蒸干,得到Na3AsS3O·8H2O晶體,溶液中存在形式:HnAsSOn-33。JP
四硫代砷酸鹽:將2.68gNa2S和1.35gNaOH溶于100mL脫氣水,加入3.48gAs2S5,加入乙醇使其沉淀。沉淀經過濾、干燥并溶解于0.2mol/LNaOH溶液,再加入乙醇再沉淀,再經過濾、干燥即得到四硫代砷酸鈉晶體(晶體化學結構式:Na3AsS4·8H2O;溶液中存在形式:HnAsS4n-3)。
上述4種晶體溶于經脫氣處理的0.1mol/LNaOH溶液中,配制各種硫代砷酸鹽標準溶液,現配現用。JP
實驗全過程在無氧氮氣環境中完成,采用即時制備的超純水,并超聲脫氣15min。
2.3儀器分析條件
電噴霧高分辨質譜(ESIHRMS)條件:流動相為甲醇0.2%乙酸(3KG-3∶KG-57,V/V),流速為0.3mL/min,進樣體積5μL,采用負離子全掃描模式,荷質比范圍m/z120~210,分辨率為70000。
離子色譜電感耦合等離子體質譜(ICICPMS)條件:IC淋洗液為80mmol/LKOH(pH12.9),流速1.3mL/min,進樣體積200μL;ICPMS功率1450W,載氣流量1.10L/min,輔助氣流量0.9L/min,監測離子m/z75,總采集時間20min。
3結果與討論
3.1ESIHRMS鑒定制備的硫代砷酸鹽
水中無機硫代砷化物單體及其脫質子產物種類繁多,詳見表1。如引言中所述,針對天然水中的砷在地表的環境地球化學過程研究,無需考慮硫代亞砷酸鹽形態,在本研究中僅合成各類硫代砷酸鹽(包括一硫代砷酸鹽、二硫代砷酸鹽、三硫代砷酸鹽和四硫代砷酸鹽)的標準樣品,采用ESIHRMS進行鑒定。JP
所制備的一硫代砷酸鹽為無色透明的短柱狀晶體,二硫代砷酸鹽為淡黃色薄片狀晶體,三硫代砷酸鹽為白色粒狀晶體,四硫代砷酸鹽為有油脂光澤白色薄片狀晶體。采用NikonUFX11生物顯微鏡及照相系統獲得的晶體照片如圖1所示。
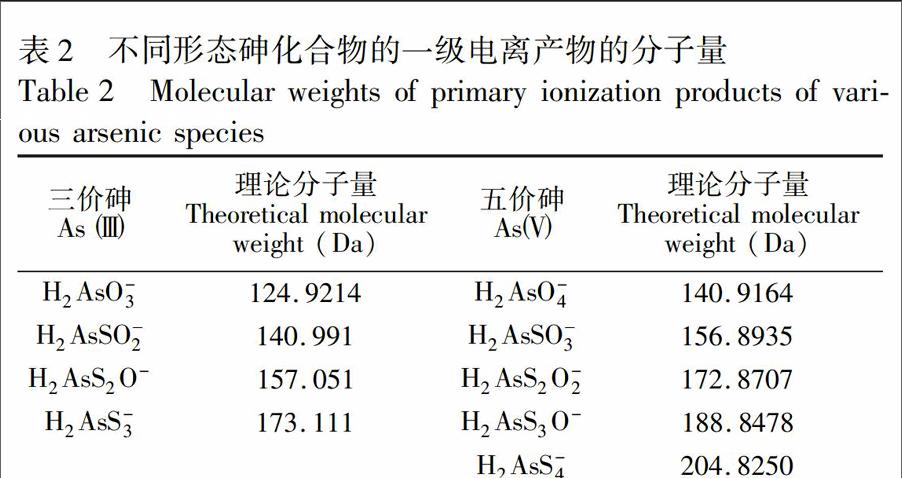
采用電噴霧高分辨質譜對合成的四種硫代砷酸鹽進行鑒定,結果如圖2所示。一硫代砷酸鹽質譜圖(圖2a)在m/z156.8931處出現唯一峰,且與H2AsSO-3理論質荷比m/z156.8935相匹配(表2),確定所合成化合物即為一硫代砷酸鹽。圖2b為二硫代砷酸鹽的譜圖,最主要質譜峰出現在m/z172.87065處,與H2AsS2O-2理論質荷比m/z172.8707一致。同理,圖2c和圖2d表明,所合成的化合物分別為三硫代砷酸鹽和四硫代砷酸鹽。圖2中各譜圖表征的化合物離子均出現了34S的同位素峰,兩個含有不同硫同位素(32S和34S)的化合物的離子質譜峰的比值與硫同位素天然豐度(32S/34S)幾乎完全一致,進一步表明合成的化合物為硫代砷酸鹽。
3.2ICICPMS分析
用ICICPMS分析了一硫代砷酸鹽、二硫代砷酸鹽、三硫代砷酸鹽、四硫代砷酸鹽、亞砷酸鹽和砷酸
鹽的單一形態溶液(As濃度50μg/L)。單一ZH(形態砷的譜圖表明:亞砷酸鹽(125s)、砷酸鹽(134s)、一硫代砷酸鹽(154s)和四硫代砷酸鹽(392s)
均可得到單一峰;二硫代砷酸鹽和三硫代砷酸鹽的主峰分別在183和268s處,但二硫代砷酸鹽譜圖同時出現3個小峰,分別為亞砷酸鹽、砷酸鹽和一硫代砷酸鹽;三硫代砷酸鹽譜圖則出現由少量砷酸鹽和一CM(19硫代、二硫代和四硫代砷酸鹽形成的雜峰。CM)ZH)上述實驗結果與文獻\[12\]的研究結果非常相似:一硫代砷酸鹽標準純度高,但其它標準均為混合物,多次結晶仍然難以進一步純化二硫代和三硫代砷酸鹽。圖3為6種形態砷化合物的混合標準物的ICICPMS譜圖,所合成的4種硫代砷酸鹽可以完全分離。下一步工作有望采用梯度洗脫分離IC系統同時分離亞砷酸鹽、砷酸鹽和硫代砷酸鹽。
3.3方法分析性能和實際樣品檢測
用ICICPMS測定了含有一定比例的亞砷酸鹽、砷酸鹽以及一硫代、二硫代、三硫代和四硫代砷酸鹽的系列濃度的混合標準溶液,除亞砷酸鹽的濃度依次為337.3,67.5,33.7,13.5,6.75和3.37μg/L外,其它標準物的濃度分別為833,166.7,83.3,33.3,16.7和8.33μg/L。結果表明(表3),總砷濃度在45.15~4515.43μg/L范圍內,除砷酸鹽由于只有一個樣品顯示出峰而無法計算相關系數外,其它標準物的相關系數R2>0.99,檢出限依次分別為0.4,25.0,3.3,1.0和1.3μg/L。表4為利用上述工作曲線測定的7個不同濃度系列的6種形態砷含量的總和與理論總砷含量的對比。所有濃度系列的濃度測定值與理論值比值在83.99%~113.60%之間,表明本方法可用于檢測總砷含量>18μg/L的水樣。對于硫代砷化物的主要天然載體——富硫化物地熱水而言,其砷含量一般在0.1~50mg/L范圍內[3\]。
將含有6種不同形態砷的混合樣品逐級稀釋,根據所測峰高或手動積分峰面積的最小濃度,以3倍標準偏差計算檢出限(表3)。
考察了所建立方法的影響因素和實際應用性能。富集硫代砷化物的天然水多見于高溫熱泉,因此,本研究選擇采自滇藏地熱帶的熱泉水樣,對其中砷的各種形態進行了測定,該熱泉樣品總砷含量為1760.1μg/L,總硫化物含量為0.21mg/L,總溶解固體為2626mg/L,水化學類型為HCO3ClNa型,主要陰陽離子分別為HCO-3、Cl
和Na+(含量分別為1241、715.9和893.4mg/L)。此外,用超純水配制了總砷含量及總硫化物含量與該熱泉樣品完全相同的水溶液,除少量Na+外,此溶液不含熱泉中的其它所有化學組分。檢測結果表明,天然熱泉樣品中砷的各種形態分別為:砷酸鹽1093.7μg/L;一硫代砷酸鹽CM(223608.2μg/L;二硫代砷酸鹽9.7μg/L;三硫代砷酸CM)
鹽48.5μg/L。配制水溶液中砷的各種形態ZH(分別為:砷酸鹽1210.4μg/L;一硫代砷酸鹽505.8μg/L;二硫代砷酸鹽8.6μg/L;三硫代砷酸鹽35.3μg/L。上述結果表明,實際天然水樣品中砷和硫化物之外的其它化學組分對硫代砷化物分析結果有一定干擾,但影響不大,可以忽略,因此,本方法具有較好的實用性。
4結論
本研究制備了4種硫代砷化物,ESICM(183HRMS鑒定結果表明所合成物質純度較CM)ZH)高,作為砷定量分析的標準物質。初步建立了基于ICICPMS聯用系統同時測定一硫代砷酸鹽、二硫代砷酸鹽、三硫代砷酸鹽、四硫代砷酸鹽、亞砷酸鹽和砷酸鹽的方法。本研究所合成的硫代砷酸鹽標準物質及其分析方法對于研究富硫化物天然水體中砷的形態及其在環境中的遷移、轉化具有一定實用意義。
References
1(#HelzGR,TossellJA.Geochim.Cosmochim.Acta,2008,72(18):4457-4468
2BostickBC,FendorfS,BrownGE.Mineral.Magaz.,2005,69(5):781-795
3KellerNS,StefánssonA,SigfússonB.Geochim.Cosmochim.Acta,2014,142:15-26
4IlgenAG,RychagovSN,TrainorTP.Chem.Geol.,2011,288(34):115-132
5RaderKJ,M.DombrowskiP,FarleyKJ,MahonyJD,ToroDMD.Environ.Toxic.Chem.,2004,23(7):1649-1654
6PlanerFredrichB,FrankeD,MerkelB,WallschlagerD.Environ.Toxic.Chem.,2008,27(10):2027-2035
7CAOXuan,YUJingJing,GAOYang,SUNJiChang,LIUYan,XUYuanYuan,SUNGuiFan,WANGXiaoRu.ChineseJ.Anal.Chem.,2012,40(1):83-88
曹煊,余晶晶,高楊,孫繼昌,劉巖,徐苑苑,孫貴范,王小如.HTK分析化學,2012,40(1):83-88
8PlanerFriedrichB,SuessE,ScheinostAC,WallschlgerD.Anal.Chem.,2010,82(24):10228-10235
9SchwedtG,RieckhoffM.J.Chromatogr.,1996,A(736):341-350
10PlanerFriedrichB,LondonJ,MccleskeyRB,NordstromDK,WallschlagerD.Environ.Sci.Technol.,2007,41:5245-5251
11KellerNS,StefanssonA,SigfussonB.Talanta,2014,128:466-472
12WallschlgerD,StadeyCJ.Anal.Chem.,2007,79(10):3873-3880
13BurtonED,BushRT,SullivanLA,JohnstonSG,HockingRK.Chem.Geol.,2008,253(12):64-73
14LaForceMJ,HanselCM,FendorfS.Environ.Sci.Technol.,2000,34(18):3937-3943
15RootRA,VlassopoulosD,RiveraNA,RaffertyMT,AndrewsC,O′DayPA.Geochim.Cosmochim.Acta,2009,73(19):5528-5553
16CoutureRM,GobeilC,TessierA.Geochim.Cosmochim.Acta,2010,74(4):1238-1255
17GuoH,LiuC,LuH,WantyRB,WangJ,ZhouY.Geochim.Cosmochim.Acta,2013,112:130-145
18SuessE,PlanerFriedrichB.Chemosphere,2012,89(11):1390-1398
19StauderS,RaueB,SacherF.Environ.Sci.Technol.,2005,39:5933-5939)
Withthesyntheticthioarsenatesasstandards,aquantitativeanalyticalmethodbasedonionchromatographinductivelycoupledplasmamassspectrometry(ICICPMS)wassetuptosimultaneouslydeterminetheconcentrationsofarsenite,arsenate,monothioarsenate,dithioarsenate,trithioarsenate,andtetrathioarsenateinwater.Thelinearrangeofthemethodwasfrom3.3μg/Lto833μg/Lwiththecorrelationcoefficientshigherthan0.99,andthedetectionlimitswerebetween0.4μg/Land25μg/L.Thesyntheticstandardmaterialsofthioarsenatesandthequantitativeanalysismethodestablishedinthisstudyareofgreatsignificanceforfutureinvestigationsontheenvironmentaltransportationandtransformationofvariousarsenicspeciesderivedfromnaturalsulfidicwaters.
KeywordsThioarsenate;Synthesis;Identification;Electrosprayionizationhighresolutionmassspectrometry;Ionchromatographinductivelycoupledplasmamassspectrometry;Quantitativeanalysis
HQWT6JY(Received18March2016;accepted30August2016)
ThisworkwassupportedbytheNationalNaturalScienceFoundationofChina(Nos.41572335,21305130)andtheResearchProgramofStateKeyLaboratoryofBiogeologyandEnvironmentalGeologyofChina(No.GBL11505).
