復合催化劑Co@Co3O4催化硼氫化鈉水解制氫性能研究*
2017-03-15 00:55:33孫丹丹謝廣文
化工科技 2017年5期
張 月,顏 雙,孫丹丹,謝廣文
(青島科技大學 材料科學與工程學院,山東 青島 266042)
經濟的高速發展使得社會對能源的需求量增加,能源過度使用、環境污染等問題越來越引起人們的重視[1-2]。為改善現有生存環境、節約化石能源、實現可持續發展,尋求一種綠色可再生能源顯得尤為重要。氫氣由于其能量密度大,燃燒產物綠色無污染,可循環使用等優點被認為是未來最有可能替代化石能源應用于生活生產的能源之一。氫氣不是一次能源,質子交換膜燃料電池可以高效地將氫能轉化為電能,由于其高效清潔的特點有望成為替代石油天然氣等化石能源的供能體[3]。然而一般情況下,氫氣都是以氣體的形式存在,其易爆易燃等特點成為阻礙實際應用的主要因素,一般的儲存方法就是高溫高壓條件下液化但成本較高[4-6]。與這種方法相比硼氫化鈉、硼氫化鉀、氨基硼烷等化學氫化物被認為是最有研究前景的儲氫材料[7-8]。在這些材料中,1 mol的硼氫化鈉可與水反應生成4 mol氫氣[9],其理論儲氫量高達10.8%,反應副產物偏硼酸鈉可以循環使用,環境友好。
貴金屬例如Pt[10-11]、Pd[12]、Ru[13-14]等,在催化堿性硼氫化鈉方面均有優異的性能,但受其價格昂貴、儲量有限的影響很難實際應用。許多研究者們將研究的重點轉向非貴金屬,Co-Ni-P[15-17],Co-P-B[18],Ni-Co-B[19],Ni-B[20]等催化劑均已被發現有良好的催化性能。金屬納米顆粒的催化性能可通過改變其組成,形貌和金屬-氧化物界面結構進行調控[21]。過渡金屬氧化物有著獨特的電子結構和磁學性能[22-23],在這些金屬氧化物中,尖晶石型四氧化三鈷擁有較大的晶胞間距、較多的活性位點[24-25]。因此作者通過不同的方法制備了納米級的鈷微粉,采用不同的氧化方法制備了Co@Co3O4復合催化劑。重點研究了液相化學還原法制備的催化劑,探討其催化堿性硼氫化鈉產氫性能,并對其結構和催化性能進行了一系列的研究。實驗表明當鈷微粉在350 ℃條件下退火制備的Co@Co3O4復合催化劑有較好的性能。
1 實驗部分
1.1 試劑與儀器
乙酸鈷、十二烷基苯磺酸鈉、次亞磷酸鈉、聚乙二醇20000:分析純,天津市巴斯夫化工有限公司;氫氧化鈉、正丁醇:分析純,雙氧水:質量分數30%,萊陽經濟技術開發區精細化工廠;氯化鈷:分析純,淄博永新化工有限公司;乙二胺:分析純,天津博迪化工股份有限公司;乙二醇:分析純,上海埃彼化學試劑有限公司;水合肼:質量分數80%,天津市廣成化學試劑有限公司;硼氫化鈉:分析純,國藥集團化學試劑有限公司。
場發射掃描電子顯微鏡:JSM-6700F,日本電子公司;X射線衍射儀:D/max-500,日本理學公司;能量色散X射線譜儀:INCA,英國Oxford儀器公司。
1.2 鈷微粉的制備
1.2.1 水熱法制備鈷微粉
稱取0.48 g CoCl2和1.2 g NaOH,量取50 mL去離子水,攪拌充分溶解;分別加入1 mL的乙二胺和水合肼,攪拌15 min;將溶液轉移到100 mL反應釜中,200 ℃下反應4 h;反應結束后自然冷卻到室溫,離心,去離子水和乙醇交替洗3次;50 ℃下真空干燥3 h,得到鈷微粉。
1.2.2 化學還原法制備鈷微粉
A液:稱取0.442 0 g Co(CH3COO)2溶于40 mL乙二醇溶液中,加入0.023 9 g十二烷基苯磺酸鈉,充分溶解。
B液:稱取1.8 g NaOH加少量水溶解,加入40 mL的乙二醇,超聲震蕩,使其混合均勻;加入0.44 g NaBH4,超聲震蕩,使其充分溶解。
用滴管將B液逐滴加入到A液中,并伴隨著超聲震蕩,滴加速度為10~15滴/min;將得到的黑色膠體放入離心機離心,水洗,醇洗各3次;50 ℃真空干燥3 h,得到鈷微粉。
1.3 Co@Co3O4復合催化劑的制備
1.3.1 退火氧化法
將制備的鈷微粉在不同溫度下退火1 h,升溫速度為4 ℃/min。在高溫狀態下,顆粒外表面與空氣充分接觸發生氧化反應生成高價態的四氧化三鈷,而顆粒內部反應不完全存在少量未被氧化的鈷微粉,經自然冷卻至室溫,得到樣品。
1.3.2 雙氧水直接氧化法
取0.5 g鈷微粉、10 mL的雙氧水分別放入到燒杯中,用一次性滴管將3~4滴雙氧水逐滴加入到鈷微粉中,不斷震蕩,直至不再有明顯氣體冒出。取吸鐵石將粉末收集,倒出反應后的液體,繼續重復上述工作,直到雙氧水耗盡。將處理好的粉末放入到烘箱中,干燥3 h,獲得樣品。
1.3.3 雙氧水水熱氧化法
取0.015 mol的鈷微粉放入燒杯A中并加入10 mL的去離子水,不斷攪拌。稱量0.78 g的聚乙二醇20000溶于37 mL的去離子水,將溶解好的聚乙二醇20000溶液倒入燒杯A中,攪拌10 min,之后加入8 mL的雙氧水和15 mL的正丁醇,攪拌30 min,放入聚四氟乙烯為內襯的水熱反應釜中,160 ℃水熱10 h,自然冷卻至室溫,離心,50 ℃干燥3 h,得到樣品。
1.4 Co@Co3O4復合催化劑催化產氫性能測試
硼氫化鈉水解制氫實驗裝置見圖1。
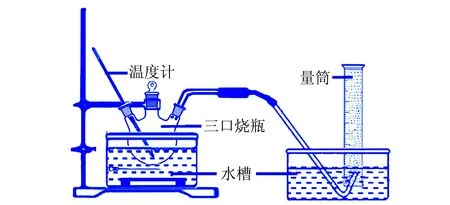
圖1 硼氫化鈉水解制氫實驗裝置圖
稱取0.8 g的硼氫化鈉溶解在20 mLw(氫氧化鈉)=10%的溶液中,將充分溶解的溶液轉移至三口燒瓶中,水浴加熱到40 ℃。準確稱量(0.010±0.001)g Co@Co3O4復合催化劑加入到三口燒瓶中,所制備的氫氣用排水法收集,用秒表計時。對時間和氫氣產生量作圖,以此表征Co@Co3O4催化劑的催化效率。
2 結果與討論
2.1 不同制備方法制備Co@Co3O4復合催化劑性能對比
2.1.1 鈷微粉的制備方法對Co@Co3O4催化劑催化產氫的影響
在w(NaBH4)=4%、w(NaOH)=10%、m(催化劑)=0.01 g、反應溫度為40 ℃條件下,不同方法合成的鈷微粉經高溫退火制備的Co@Co3O4催化劑的催化性能見圖2。
從圖2可以看出,由水熱法制備的鈷微粉氧化后得到的復合催化劑在催化體系中響應時間較長,化學還原法制備催化劑反應迅速,響應時間較短。從熱力學上分析,化學還原法制備的催化劑屬于非晶結構處于亞穩態[26],能量較高更容易催化硼氫化鈉水解釋放氫氣。采用雙氧水直接氧化、雙氧水水熱氧化兩種方法制備的Co@Co3O4復合催化劑同樣證明化學還原法制備的鈷微粉性能較好,因此選用化學還原法制備的鈷微粉進一步研究。
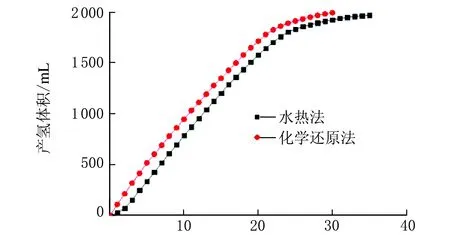
t/min圖2 鈷微粉的制備方法對Co@Co3O4 催化劑催化產氫的影響
2.1.2 氧化方法對Co@Co3O4復合催化劑的催化產氫的影響
在w(NaBH4)=4%、w(NaOH)=10%、m(催化劑)=0.01 g、反應溫度為40 ℃條件下,不同的氧化方法制備Co@Co3O4復合催化劑的催化性能對比見圖3。

t/min圖3 氧化方法對Co@Co3O4復合催化劑催化產氫的影響
從圖3可以看出,經雙氧水氧化后的樣品產氫性能低于鈷微粉。350 ℃退火制備的Co@Co3O4復合催化劑性能優于鈷單質以及其它的氧化方法制備的Co@Co3O4復合催化劑。說明采用退火方式制備的Co@Co3O4催化劑具有較好的性能,進一步探究不同退火溫度對Co@Co3O4復合催化劑性能的影響。
2.1.3 退火溫度對Co@Co3O4復合催化劑催化產氫的影響
在w(NaBH4)=4%、w(NaOH)=10%、m(催化劑)=0.01 g、反應溫度為40 ℃條件下,不同退火溫度制備的Co@Co3O4復合催化劑催化堿性硼氫化鈉產氫性能見圖4。

t/min圖4 退火溫度對Co@Co3O4復合催化劑催化產氫的影響
由圖4可知,退火溫度對催化性能有較大的影響。退火溫度為350 ℃時,催化劑活性顯著提高。當t=15 min時,產氫體積約為1 650 mL,其催化硼氫化鈉溶液的平均產氫速率可達11.0 L/(g·min)。這明顯高于Eom K S等[27]通過化學鍍法制備的Co-P催化劑和李等[16]通過化學鍍法制備的Co-Ni-P/γ-Al2O3催化劑。這主要是由于以下兩方面的原因。第一,該催化劑獨特的非晶結構。隨退火溫度的升高,催化劑逐漸晶化,催化活性降低,因而催化速率下降。李等證明非晶態結構的催化劑催化效率明顯要比晶態結構的要高[27]。第二,合理的催化劑成分組成。不同溫度退火后,催化劑復合結構中Co3O4和Co的含量會出現明顯差異,這會對催化劑的催化性能產生顯著影響。
2.2 Co@Co3O4催化劑的微觀形貌、元素分析與晶體結構
2.2.1 Co@Co3O4催化劑的微觀形貌
鈷微粉與不同退火溫度制備的Co@Co3O4復合催化劑SEM照片見圖5。
圖5a為采用化學還原法制備的鈷微粉,圖5b~5d為鈷微粉(圖5a)經不同溫度退火1h得到的Co@Co3O4復合催化劑。由圖5a可知,化學還原法制備的鈷微粉粒徑均一,直徑小于150 nm。如圖5b所示,當退火溫度為350 ℃時,制備的催化劑粒徑較小(約200 nm)。由圖5b~5d可得,隨著退火溫度的增加,催化劑顆粒粒徑不斷增加,小顆粒團聚逐漸長成形狀不規則的大顆粒,形貌迥異。一定程度上解釋了在高溫區隨退火溫度的增加催化劑活性下降這一現象。

a 鈷微粉

b 350 ℃退火制備Co@Co3O4催化劑

c 400 ℃退火制備Co@Co3O4催化劑

d 450 ℃退火制備Co@Co3O4催化劑圖5 鈷微粉與不同氧化方法制備的Co@Co3O4 復合催化劑SEM照片
由圖5可知,通過化學還原法制備的鈷微粉粒徑較小接近納米級別。350 ℃退火處理使得部分催化劑在晶化的同時氧化成Co3O4,使其催化性能增強,而且該催化劑粒徑變化不明顯,結晶化程度不高,保留了非晶態催化劑獨特的短程有序、長程無序的特點,賦予它更高的表面活性中心密度。
2.2.2 Co@Co3O4催化劑的元素分析
化學還原法制備的鈷微粉經350 ℃退火合成的Co@Co3O4復合催化劑EDS譜圖見圖6。

E/keV圖6 Co@Co3O4復合催化劑的EDS譜圖
從譜圖上可以清晰的找到鈷、氧、碳各元素的特征峰,表明成功制備了Co@Co3O4復合催化劑。其中碳的峰較高,這主要是測試時所使用的導電膠和反應過程中加入的表面活性劑十二烷基苯磺酸鈉共同作用的結果。
Co@Co3O4復合催化劑在電子束激發情況下所產生的特征X射線中的K線系譜線的EDS分析結果見表1。

表1 Co@Co3O4復合催化劑的EDS分析結果
2.2.3 Co@Co3O4催化劑的晶體結構
鈷微粉和Co@Co3O4復合催化劑的XRD譜圖見圖7。

2θ/(°)圖7 不同樣品的XRD譜圖
從圖7a可以看出,化學還原法制備的鈷微粉的X射線衍射在2θ≈20°、45°附近各有一個寬泛的特征彌散峰,這是非晶態結構比較典型的特征峰,說明合成的鈷微粉的物相結構為非晶態。由圖7b~7d可以看出,退火處理后出現了明顯的衍射峰,并隨退火溫度的升高,衍射峰逐漸增強。根據分析可知,溫度越高,氧化生成的Co3O4含量越高,說明在空氣中退火處理,鈷微粉顆粒外表面逐漸氧化變成高價態的Co3O4并逐漸向結晶態轉化,形成由鈷微粉和氧化生成的Co3O4并存的非晶與微晶復合結構[28]。這是由于非晶態合金在熱力學上處于亞穩態,在一定溫度下退火可以使催化劑結構從非晶態轉變為晶態[29]。衍射峰的2θ值分別為31.271°、36.852°、59.357°、65.236°,對應標準卡片(JCPDS,No.42-1467)結晶態的立方相Co3O4(220)、(311)、(511)、(440)晶面。2θ為41.683°、44.762°處的衍射峰對應標準卡片(JCPDS,No.05-0727)六方結構鈷的(100)、(002)晶面。
2.3 Co@Co3O4復合催化劑磁性能測試
取適量Co@Co3O4復合催化劑加少量水超聲分散,將磁鐵置于燒杯外壁,Co@Co3O4復合催化劑在磁鐵作用下的現象見圖8。

圖8 Co@Co3O4復合催化劑在磁鐵作用下的現象
由圖8可見,分散在水中的顆粒迅速聚集在一起并可以根據磁鐵的運動而運動。鈷是鐵磁性材料,有較強的磁性,受鈷的影響,復合催化劑擁有較強的磁性,利于分離收集回用。
3 結 論
通過化學還原法,以硼氫化鈉為還原劑在室溫下制備納米鈷微粉,在350 ℃條件下退火氧化得到粒徑較小的Co@Co3O4復合催化劑。研究了Co@Co3O4復合催化劑催化水解硼氫化鈉的速率與鈷微粉的制備方法,氧化方式的關系。探究了退火溫度對Co@Co3O4復合催化劑催化硼氫化鈉水解制氫性能的影響。結果表明,液相化學還原法制備的鈷微粉經退火后所得Co@Co3O4復合催化劑擁有較高的催化活性,在40 ℃催化硼氫化鈉水解產氫速率為11.0 L/(g·min),并且具有一定的磁性,便于反應后催化劑的分離和收集,這在一定程度上克服了粉末狀樣品難回收的缺點。
[1] LUND H,MATHIESEN B V.Energy system analysis of 100% renewable energy systems——the case of Denmark in years 2030 and 2050[J].Energy,2009,34(5):524-531.
[2] FERNANDES R,PATEL N,MIOTELLO A.Hydrogen generation by hydrolysis of alkaline NaBH4solution with Cr-promoted Co-B amorphous catalyst[J].Applied Catalysis B:Environmental,2009,92(1):68-74.
[3] JIANG G M,LI X W,LV X S,et al.Core/shell FePd/Pd catalyst with a superior activity to Pt in oxygen reduction reaction[J].Science Bulletin,2016,61(16):1248-1254.
[4] SANTOS D M F,SEQUEIRA C A C.Sodium borohydride as a fuel for the future[J].Renewable and Sustainable Energy Reviews,2011,15(8):3980-4001.
[5] AHLUWALIA R K,HUA T Q,PENG J K.Fuel cycle efficiencies of different automotive on-board hydrogen storage options[J].International Journal of Hydrogen Energy,2007,32(15):3592-3602.
[6] WANG W L,ZHAO Y C,CHEN D H,et al.Promoted Mo incorporated Co-Ru-B catalyst for fast hydrolysis of NaBH4in alkaline solutions[J].International Journal of Hydrogen Energy,2014,39(28):16202-16211.
[7] ZHAO Y C,NING Z,TIAN J N,et al.Hydrogen generation by hydrolysis of alkaline NaBH4solution on Co-Mo-Pd-B amorphous catalyst with efficient catalytic properties[J].Journal of Power Sources,2012,207:120-126.
[8] PENA ALONSO R,SICURELLI A,CALLONE E,et al.A picoscale catalyst for hydrogen generation from NaBH4for fuel cells[J].Journal of Power Sources,2007,165(1):315-323.
[9] JENA P.Materials for hydrogen storage:past,present,and future[J].The Journal of Physical Chemistry Letters,2011,2(3):206-211.
[10] XU D Y,ZHANG H M,YE W.Hydrogen generation from hydrolysis of alkaline sodium borohydride solution using Pt/C catalyst[J].Catalysis Communications,2007,8(11):1767-1771.
[11] BAI Y,WU C,WU F,et al.Carbon-supported platinum catalysts for on-site hydrogen generation from NaBH4solution[J].Materials Letters,2006,60(17):2236-2239.
[12] GUELLA G,ZANCHETTA C,PATTON B,et al.New insights on the mechanism of palladium-catalyzed hydrolysis of sodium borohydride from 11 B NMR measurements[J].The Journal of Physical Chemistry B,2006,110(34):17024-17033.
[13] HUANG Y H,SU C C,WANG S L,et al.Development of Al2O3carrier-Ru composite catalyst for hydrogen generation from alkaline NaBH4hydrolysis[J].Energy,2012,46(1):242-247.
[14] ZAHMAKIRAN M,OZKAR S.Zeolite-confined ruthenium(0)nanoclusters catalyst:record catalytic activity,reusability,and lifetime in hydrogen generation from the hydrolysis of sodium borohydride[J].Langmuir,2009,25(5):2667-2678.
[15] GUO Y P,FENG Q H,MA J T.The hydrogen generation from alkaline NaBH4solution by using electroplated amorphous Co-Ni-P film catalysts[J].Applied Surface Science,2013,273(15):253-256.
[16] LI Z,LI H L,WANG L N,et al.Hydrogen generation from catalytic hydrolysis of sodium borohydride solution using supported amorphous alloy catalysts (Ni-Co-P/γ-Al2O3)[J].International Journal of Hydrogen Energy,2014,39(27):14935-14941.
[17] KIM D R,CHO K W,CHOI Y,et al.Fabrication of porous Co-Ni-P catalysts by electrodeposition and their catalytic characteristics for the generation of hydrogen from analkaline NaBH4solution[J].International Journal of Hydrogen Energy,2009,34(6):2622-2630.
[18] PATEL N,FERNANDES R,BAZZANELLA N,et al.Co-P-B catalyst thin films prepared by electroless and pulsed laser deposition for hydrogen generation by hydrolysis of alkaline sodium borohydride:a comparison[J].Solid Films,2010,518(17):4779-4785.
[19] XU D Y,WANG H Z,GUO Q J,et al.Catalytic behavior of carbon supported Ni-B,Co-B and Co-Ni-B in hydrogen generation by hydrolysis of KBH4[J].Fuel Process Technology,2011,92(8):1606-1610.
[20] LEE J K,ANN H H,YI Y,et al.A stable Ni-B catalyst in hydrogen generation via NaBH4hydrolysis[J].Catalysis Communications,2011,16(1):120-123.
[21] WHITE R J,LUQUE R,BUDARIN V L,et al.Supported metal nanoparticles on porous materials.Methods and applications[J].Chemical Society Reviews,2009,38(2):481-494.

[23] CHAE J,PYAGAI R,DEMIDIOUK V,et al.Influence of cobalt precursors on the catalytic activity of the cobalt oxide/Al2O3catalysts[J].Reaction Kinetics and Catalysis Letters,2004,83(2):369-375.
[24] LARCHER D,SUDANT G,LERICHE J B,et al.The electrochemical reduction of Co3O4in a lithium cell[J].Journal of the Electrochemical Society,2002,149(3):A234-A241.
[25] HAN B,PARK J M,CHOI K H,et al.Atomic layer deposition of stoichiometric Co3O4films using bis (1,4-di-iso-propyl-1,4-diazabutadiene) cobalt[J].Thin Solid Films,2015,589(31):718-722.
[26] 王麗娜,李忠,劉騰宇,等.負載型非晶合金催化劑 Ni-Co-P/γ-Al2O3的制備及其結構和催化性能表征[J].青島科技大學學報(自然科學版),2016,37(1):41-46.
[27] EOM K S,CHO K W,KWON H S.Effects of electroless deposition conditions on microstructures of cobalt-phosphorous catalysts and their hydrogen generation properties in alkaline sodium borohydride solution[J].Journal of Power Sources,2008,180(1):484-490.
[28] 鐘明龍.Co3O4納米片的簡單熱氧化法制備及其磁性能[J].稀有金屬,2015,39(2):119-123.
[29] 李忠,趙興蕾,王麗娜,等.負載型非晶合金催化劑Ni-Co-P/γ-Al2O3催化硼氫化鈉水解制氫[J].青島科技大學學報(自然科學版),2014,35(6):582-586.
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
科技知識動漫(2017年7期)2017-08-09 19:52:45
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
科技知識動漫(2016年10期)2016-10-18 20:35:00
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
