和胃顆粒質(zhì)量標(biāo)準(zhǔn)研究
2017-01-05 03:56:14廖格斯于士龍趙慶春
實(shí)用藥物與臨床 2016年12期
廖格斯,汪 宇,于士龍,趙慶春*
和胃顆粒質(zhì)量標(biāo)準(zhǔn)研究
廖格斯1,汪 宇2,于士龍2,趙慶春1*
目的 建立和胃顆粒的質(zhì)量標(biāo)準(zhǔn)。方法 采用TLC法定性鑒別處方中半夏、甘草、人參、黃連、干姜含量;采用HPLC法測(cè)定黃芩苷的含量。色譜柱為Agilent TC-C18(4.6 mm×250 mm,5 μm);以乙腈為流動(dòng)相A、0.1%磷酸溶液為流動(dòng)相B,按規(guī)定進(jìn)行梯度洗脫;檢測(cè)波長(zhǎng)為280 nm;流速1.0 mL/min;柱溫25 ℃。結(jié)果 5種成分的薄層定性鑒別分離、專(zhuān)屬性均較好;黃芩苷在0.070 26~1.053 86 μg范圍內(nèi)線性關(guān)系良好(r=0.999 9),平均加樣回收率為99.7%,RSD為1.01% (n=6)。結(jié)論 本方法可以較好地控制該制劑的質(zhì)量。
和胃顆粒;質(zhì)量標(biāo)準(zhǔn);TLC;HPLC
0 引言
和胃顆粒是中國(guó)人民解放軍第四六三醫(yī)院生產(chǎn)的醫(yī)院制劑,由法半夏、生姜、茯苓、人參、黃芩、甘草、黃連等十余味藥材組成,具有益氣和胃、消痞散結(jié)的功效。主要用于放、化療引起的惡心、嘔吐、食欲不振,也可用于慢性胃炎、胃動(dòng)力不足、胃潰瘍等疾病。在臨床上有很大的需求。目前和胃顆粒的質(zhì)量標(biāo)準(zhǔn)中,薄層鑒別藥味少,且無(wú)含量測(cè)定項(xiàng)目。為了更好地控制該制劑的質(zhì)量,筆者采用薄層色譜法對(duì)制劑中干姜、甘草、人參、黃芩、半夏進(jìn)行定性鑒別,采用HPLC法測(cè)定處方中主要成分黃芩中指標(biāo)成分黃芩苷的含量[1-2],對(duì)質(zhì)量標(biāo)準(zhǔn)進(jìn)行了完善,現(xiàn)報(bào)道如下。
1 儀器與試藥
高效液相色譜儀(日本島津LC-2010AHT);KQ3200E型昆山超聲波處理器;電子分析天平(120D型,北京島津公司)。黃芩苷對(duì)照品(中國(guó)食品藥品檢定研究院,供含量測(cè)定用,含量95.2%,批號(hào):110715-200815);和胃顆粒(中國(guó)人民解放軍第四六三醫(yī)院提供,批號(hào):20140422、20140603、20140915);半夏對(duì)照藥材(批號(hào):121272-201404)、甘草對(duì)照藥材(批號(hào):120904-201318)、人參對(duì)照藥材(批號(hào):120904-201318)、黃連對(duì)照藥材(批號(hào):120913-201310)、干姜對(duì)照藥材(批號(hào):120942-201309),均購(gòu)自中國(guó)食品藥品檢定研究院;乙腈(色譜純,Tedia公司),水為純化水,其他試劑均為分析純。
2 方法與結(jié)果
2.1 半夏薄層鑒別 取本品5 g,研細(xì),加95%乙醇50 mL,超聲處理20 min,濾過(guò),濾液置水浴鍋上揮發(fā)溶劑至剩余約2 mL,作為供試樣品液;根據(jù)本制劑中各種藥材比例(半夏除外),按制備工藝制成顆粒,按供試樣品液制備方法制成陰性對(duì)照液;取半夏對(duì)照藥材1 g,加95%乙醇20 mL,置超聲波處理器中超聲20 min,蒸干后的殘?jiān)右宜嵋阴? mL溶解,作為對(duì)照藥材溶液。分別用定量點(diǎn)樣器在同一硅膠G薄層板上點(diǎn)樣上述3種溶液各10 μL,于層析缸中展開(kāi)[展開(kāi)劑為正丁醇-冰醋酸-水(8∶3∶1)],至展距16 cm取出,揮干板上殘留溶劑后,噴以顯色劑(茚三酮試液),置烘箱中105 ℃加熱約5 min。3批樣品的供試樣品斑點(diǎn)與半夏對(duì)照藥材相應(yīng)的位置上,顯相同顏色的斑點(diǎn)。陰性對(duì)照無(wú)干擾。見(jiàn)圖1。

圖1 半夏薄層色譜鑒別
2.2 甘草薄層鑒別 取本品5 g,研細(xì),加甲醇50 mL,超聲處理20 min,濾液置水浴鍋上揮發(fā)溶劑至剩余約2 mL,作為供試樣品液;根據(jù)本制劑中各種藥材比例(甘草除外),按制備工藝制成顆粒,按供試樣品液制備方法制成陰性對(duì)照液;取甘草對(duì)照藥材1 g,加甲醇20 mL,置超聲波處理器中超聲20 min,置水浴鍋上揮發(fā)溶劑至剩余約2 mL,作為甘草對(duì)照藥材溶液。分別用定量點(diǎn)樣器在同一硅膠G薄層板上點(diǎn)樣上述3種溶液各10 μL,于層析缸中展開(kāi)[展開(kāi)劑為乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)],至展距16 cm取出,揮干板上殘留溶劑后,噴以顯色劑(10%硫酸乙醇溶液),置烘箱中105 ℃加熱約5 min。3批樣品的供試樣品斑點(diǎn)與甘草對(duì)照藥材相應(yīng)的位置上,顯相同顏色的斑點(diǎn)。陰性對(duì)照無(wú)干擾。見(jiàn)圖2。

圖2 甘草薄層色譜鑒別
2.3 人參薄層鑒別 取本品3 g,研細(xì),加入100 mL甲醇,置水浴鍋內(nèi)回流提取3 h,放冷,蒸至近干的濾液加水至20 mL,用水飽和正丁醇提取(共3次,20 mL/次),合并提取液,用氨試液洗滌(共3次,20 mL/次),棄去氨試液,再用正丁醇飽和的水溶液洗滌(共2次,20 mL/次),取洗滌液蒸至近干后,加水至30 mL,得濾液,置水浴鍋上揮發(fā)溶劑至約5 mL,通過(guò)D101大孔吸附樹(shù)脂柱(內(nèi)徑1.5 cm,柱高12 cm)先以水洗至流出液近無(wú)色,用400 mL 40% 乙醇溶液洗脫至無(wú)色,繼續(xù)用70%乙醇溶液180 mL洗脫并收集洗脫液,水浴揮干溶劑后用1 mL甲醇溶解,作為供試樣品溶液;根據(jù)本制劑中各種藥材比例(除人參外),按制備工藝制成顆粒,按供試樣品溶液操作方法制成陰性對(duì)照溶液;另取人參對(duì)照藥材1 g,同供試樣品溶液方法制成對(duì)照藥材溶液。分別用定量點(diǎn)樣器在同一硅膠G薄層板上點(diǎn)樣上述3種溶液各10 μL,于層析缸中展開(kāi)[展開(kāi)劑為二氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)],至展距16 cm取出,揮干板上殘留溶劑后,噴以顯色劑(10%硫酸乙醇溶液),置烘箱中105 ℃加熱約5 min。3批樣品的供試樣品斑點(diǎn)與人參對(duì)照藥材相應(yīng)的位置上,顯相同顏色的斑點(diǎn)。陰性對(duì)照無(wú)干擾。見(jiàn)圖3。

圖3 人參薄層色譜鑒別
2.4 黃連薄層鑒別 取本品10 g,研細(xì),加95%乙醇50 mL,超聲處理20 min,濾液置水浴鍋上揮發(fā)溶劑至剩余約2 mL,作為供試樣品液;根據(jù)本制劑中各種藥材比例(黃連除外),按制備工藝制成顆粒,按供試樣品液制備方法制成陰性對(duì)照液;取黃連對(duì)照藥材1 g,加95%乙醇20 mL,置超聲波處理器中超聲20 min,濾液置水浴鍋上揮發(fā)溶劑至剩余約2 mL,作為對(duì)照藥材溶液。分別用定量點(diǎn)樣器在同一硅膠G薄層板上點(diǎn)樣上述3種溶液各10 μL,再用濃氨試液預(yù)飽和20 min的展缸內(nèi)展開(kāi)[展開(kāi)劑為環(huán)己烷-乙酸乙酯-異丙醇-甲醇-水-三乙胺(3∶3.5∶1∶1.5∶0.5∶1)],至展距16 cm取出,揮干板上殘留溶劑后,在365 nm紫外燈下檢視。3批樣品的供試樣品斑點(diǎn)與黃連對(duì)照藥材相應(yīng)的位置上,顯相同顏色的斑點(diǎn)。陰性對(duì)照無(wú)干擾。見(jiàn)圖4。

圖4 黃連薄層色譜鑒別
2.5 干姜薄層鑒別 取本品10 g,研細(xì),加95%乙醇50 mL,超聲處理20 min,濾液置水浴鍋上揮發(fā)溶劑至剩余約2 mL,作為供試樣品液;根據(jù)本制劑中各種藥材比例(干姜除外),按制備工藝制成顆粒,按供試樣品液制備方法制成陰性對(duì)照液;干姜對(duì)照藥材0.5 g,加95%乙醇20 mL,置超聲波處理器中超聲20 min,濾液置水浴鍋上揮發(fā)溶劑至剩余約2 mL,作為干姜對(duì)照藥材溶液。分別用定量點(diǎn)樣器在同一硅膠G薄層板上點(diǎn)樣上述3種溶液各10 μL,再用濃氨試液預(yù)飽和20 min的展缸內(nèi)展開(kāi)[展開(kāi)劑為環(huán)己烷∶乙酸乙酯(1∶1)],至展距16 cm取出,噴以顯色劑(香草醛硫酸試液),置烘箱中105 ℃加熱約5 min。3批樣品的供試樣品斑點(diǎn)與干姜對(duì)照藥材相應(yīng)的位置上,顯相同顏色的斑點(diǎn)。陰性對(duì)照無(wú)干擾。見(jiàn)圖5。

圖5 干姜薄層色譜鑒別
2.6 黃芩苷含量測(cè)定
2.6.1 色譜條件 色譜柱:Agilent TC-C18(4.6 mm×250 mm,5 μm);流動(dòng)相:以乙腈為流動(dòng)相A,以0.1%磷酸溶液為流動(dòng)相B,按表1的比例梯度洗脫;檢測(cè)波長(zhǎng)為 280 nm;流速1.0 mL/min;柱溫25 ℃。
2.6.2 溶液的制備 黃芩苷對(duì)照品溶液的制備:精密稱(chēng)取黃芩苷對(duì)照品適量,加70%乙醇制成每1 mL含15 μg的溶液,即得。供試品溶液的制備:精密稱(chēng)取事先研細(xì)的本品(0.2 g),置25 mL的量瓶中,精密加入70%乙醇25 mL,密閉,稱(chēng)量。超聲處理30 min,放冷,稱(chēng)重,加入70%乙醇至超聲處理前溶液的重量,搖勻,濾過(guò),即得。陰性對(duì)照樣品溶液的制備:根據(jù)本制劑中各種藥材比例(黃芩除外),按制備工藝制成顆粒,再按供試品溶液的制備方法制得陰性樣品溶液。色譜圖見(jiàn)圖6。

表1 梯度洗脫條件
2.6.3 標(biāo)準(zhǔn)曲線的制備 精密稱(chēng)取黃芩苷對(duì)照品適量,加70%乙醇制成每1 mL含0.035 13 mg的溶液,分別精密吸取該對(duì)照品溶液2、5、10、15、20、30 μL,進(jìn)樣,按“2.6.1”項(xiàng)色譜條件測(cè)定6種不同濃度溶液峰面積,得回歸方程(以進(jìn)樣量為橫坐標(biāo)、峰面積積分值為縱坐標(biāo)作圖)為Y=2 745.1X-29 859,r=0.999 9。結(jié)果表明,黃芩苷對(duì)照品量與測(cè)得的峰面積呈良好的線性關(guān)系(0.070 26~1.053 86 μg)。
2.6.4 黃芩苷對(duì)照品精密度試驗(yàn) 精密吸取含對(duì)照品的溶液(0.035 13 mg/μL),按“2.6.1”項(xiàng)方法連續(xù)測(cè)定6次,測(cè)得峰面積值的RSD為0.33% (n=6),符合要求。
2.6.5 穩(wěn)定性 取同一批樣品(批號(hào):20140522),按照“2.6”項(xiàng)下方法和條件進(jìn)行分析,分別在0、4、8、12、16、24 h測(cè)定樣品中黃芩苷峰面積,RSD為0.18% (n=6),表明供試品溶液在24 h內(nèi)穩(wěn)定。
2.6.6 重復(fù)性試驗(yàn) 取同一批樣品(批號(hào):20140422),按照“2.6”項(xiàng)下方法和條件進(jìn)行制備及分析,測(cè)定每份溶液含量為平均標(biāo)示量的99.8%,RSD為0.33% (n=6),符合要求。
2.6.7 加樣回收率試驗(yàn) 精密稱(chēng)取和胃顆粒(批號(hào):20140422,黃芩苷含量為2.145 7 mg/g) 0.08、0.1、0.12 g 3個(gè)梯度樣品,每個(gè)梯度3份,分別加入25 mL量瓶中;稱(chēng)取黃芩苷對(duì)照品(含量:95.2%) 5.96 mg,置100 mL量瓶中,70%乙醇定容至刻度,再分別取3.2、4.0、4.8 mL對(duì)照品溶液加入上述已稱(chēng)取和胃顆粒的3個(gè)梯度量瓶中,再加70%乙醇定容至刻度,密閉,稱(chēng)量。超聲處理30 min,放冷,稱(chēng)重,用70%乙醇補(bǔ)足減失的重量,搖勻,濾過(guò),即得,按“2.6”項(xiàng)下供試樣品溶液方法和條件進(jìn)行分析,其平均回收率為99.6%,RSD為1.01% (n=9),結(jié)果見(jiàn)表2。
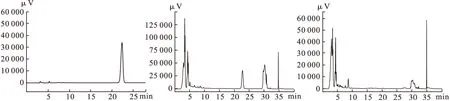
圖6 和胃顆粒色譜圖
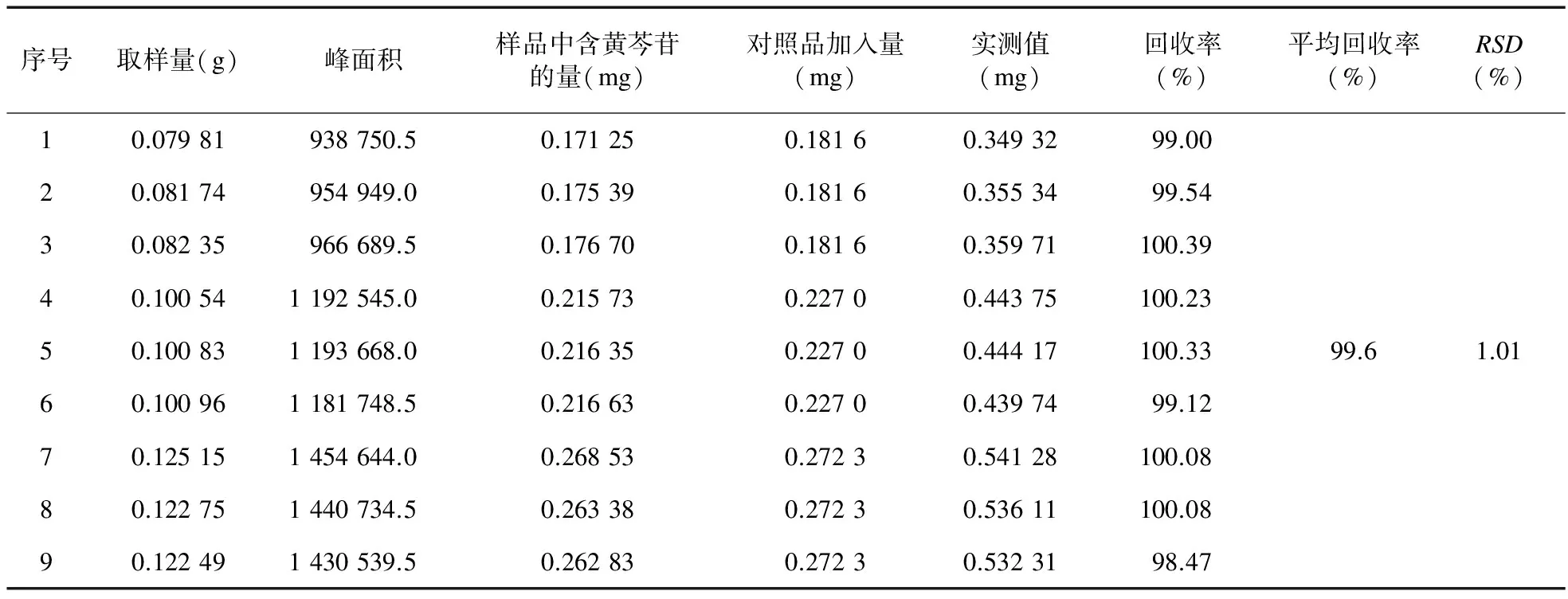
序號(hào)取樣量(g)峰面積樣品中含黃芩苷的量(mg)對(duì)照品加入量(mg)實(shí)測(cè)值(mg)回收率(%)平均回收率(%)RSD(%)10.07981938750.50.171250.18160.3493299.0020.08174954949.00.175390.18160.3553499.5430.08235966689.50.176700.18160.35971100.3940.100541192545.00.215730.22700.44375100.2350.100831193668.00.216350.22700.44417100.3399.61.0160.100961181748.50.216630.22700.4397499.1270.125151454644.00.268530.27230.54128100.0880.122751440734.50.263380.27230.53611100.0890.122491430539.50.262830.27230.5323198.47
2.6.8 樣品測(cè)定 取3個(gè)批號(hào)樣品,按“2.6.2”項(xiàng)下供試品溶液制備操作,按該項(xiàng)下色譜條件測(cè)定各自樣品中黃芩苷的含量。結(jié)果見(jiàn)表3。

表3 三批樣品含量測(cè)定結(jié)果(mg/g,n=2)
3 討論
3.1 干姜定性鑒別的優(yōu)化 供試品溶液的制備中,筆者曾嘗試采用2種方法提取:①用甲醇超聲提取;②用95%乙醇超聲提取。結(jié)果顯示,2種方法薄層斑點(diǎn)都較清晰,考慮甲醇毒性較大,故將乙醇提取納入正文。展開(kāi)系統(tǒng)藥典方法為石油醚-三氯甲烷-乙酸乙酯(2∶1∶1),但三氯甲烷毒性大[3],本文嘗試選取環(huán)己烷-乙酸乙酯(1∶1)為展開(kāi)劑,分離效果好,取3批樣品試驗(yàn),經(jīng)過(guò)考查,該方法斑點(diǎn)清晰,重現(xiàn)性好,而且穩(wěn)定,陰性無(wú)干擾,故將此方法列入正文。
3.2 提取方法考察 分別對(duì)溶劑用量、提取時(shí)間進(jìn)行考察。溶劑用量考察:分別使用70%乙醇25、50、100 mL提取,黃芩苷的含量測(cè)定結(jié)果無(wú)顯著性差異,為降低成本、減少污染,選用25 mL作為提取溶劑用量。提取時(shí)間考察:樣品分別超聲提取20、30、40 min,提取30、40 min時(shí)黃芩苷的提取率相近,且高于20 min時(shí)的提取率,故將提取時(shí)間定為30 min。
3.3 梯度洗脫條件考察 “2.6”項(xiàng)中,選擇流動(dòng)相時(shí),根據(jù)中國(guó)藥典[4]和相關(guān)文獻(xiàn)[5-6]選擇了甲醇-0.05%磷酸水溶液(三乙胺調(diào)pH值為3.0)梯度洗脫、甲醇-0.2%磷酸水(45∶55)及甲醇-水-磷酸(47∶53∶0.2)為流動(dòng)相進(jìn)行考察,結(jié)果分離度及峰形均不理想,最后選擇表1的梯度洗脫條件,黃芩苷的峰形較好,測(cè)定分離度符合要求。
[1] 張瑜,武斌,許建衛(wèi),等.黃芩藥理作用的研究進(jìn)展[J].醫(yī)學(xué)綜述,2013,19(6):1091-1093.
[2] 劉征輝,魏靜娜,趙琳琳,等.黃芩提取物多指標(biāo)成分鑒定及指紋圖譜的研究[J].世界科學(xué)技術(shù):中醫(yī)藥現(xiàn)代化,2015,17(1):156-161.
[3] 王子友.三氯甲烷職業(yè)中毒的預(yù)防[J].勞動(dòng)保護(hù),2010,(8):84-85.
[4] 國(guó)家藥典委員會(huì).中華人民共和國(guó)藥典(一部)[S].北京:中國(guó)醫(yī)藥科技出版社,2010:542-543.
[5] 張彥麗,王艷,李新霞,等.高效液相色譜法測(cè)定昆侖雪菊中綠原酸和黃芩苷的含量[J].中國(guó)實(shí)驗(yàn)方劑學(xué)雜志,2012,18(4):107-109.
[6] 宋亞芳,毛娜,楊紅,等.高效液相色譜法測(cè)定銀黃顆粒中黃芩苷的含量[J].中國(guó)實(shí)驗(yàn)方劑學(xué)雜志,2011,17(15):99-100.
Quality standards for Hewei granules
LIAO Ge-si1,WANG Yu2,YU Shi-long2,ZHAO Qing-chun1*
(1.Shenyang Pharmaceutical University,Shenyang 110016,China;2.NO.463 Hospital of PLA,Shenyang 110042,China)
Objective To establish the quality standard for Hewei granules.MethodsPinelliaternata,Liquorice,Ginseng,CoptischinensisandRhizomazingiberiswere identified by TLC and the contents of baicalin were determined by RP-HPLC.The chromatography conditions were as follows:Agilent TC-C18column (4.6 mm×250 mm,5 μm);the mobile phase consisted of A (acetonitrile) and B (0.1% phosphoric acid) with the column temperature being 25 ℃ and gradient elution;UV detection wavelength was set at 280 nm with the flow rate of 1.0 mL/min.The sample injection was 10 μL.Results The method of TLC had good resolution with clear spots,and negative control showed no interference.The good linearity was obtained within the range of 0.070 26~1.053 86 μg (r=0.999 9) for baicalin,and the average recovery was 99.7% (RSD=1.01%,n=6).Conclusion The established method is simple,accurate and sensitive for the the quality control of Hewei granules.
Hewei granules;Quality standards;TLC;HPLC
2016-05-12
1.沈陽(yáng)藥科大學(xué),沈陽(yáng) 110016;2.沈陽(yáng)軍區(qū)第四六三醫(yī)院,沈陽(yáng)110042
*通信作者
10.14053/j.cnki.ppcr.201612020
