Cu/Mn/Ce三元氧化物催化劑對甲苯催化燃燒性能的研究
2016-11-15 11:55:06朱紅娜呂德義郇昌永
浙江化工 2016年10期
關鍵詞:催化劑
朱紅娜,呂德義,郇昌永
(1.浙江工業大學化學工程學院,浙江杭州310014;2.寧波化學工業區博士后科研工作站,浙江寧波315204)
Cu/Mn/Ce三元氧化物催化劑對甲苯催化燃燒性能的研究
朱紅娜1,呂德義1,郇昌永2*
(1.浙江工業大學化學工程學院,浙江杭州310014;2.寧波化學工業區博士后科研工作站,浙江寧波315204)
采用浸漬法制備了不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑。通過XRD、和H2-TPR對催化劑的晶相結構和晶格氧的移動性進行了表征和分析,采用常壓氣-固反應裝置考察了不同的銅錳鈰摩爾比對催化氧化甲苯性能的影響,并討論了催化劑晶相組成、氧移動性與催化活性的關系。結果顯示,銅錳氧化物復合形成的新相Cu1.4Mn1.6O4是具有更高催化活性的中心,CeO2的加入提高了Cu1.4Mn1.6O4分散度,從而增強了催化劑的氧移動性,提高了催化劑的活性和氧化能力。當Cu/Mn/Ce摩爾比為1:1:4時,催化氧化甲苯的活性最佳,T95%為282.6℃,CO2選擇性為100%。
銅錳鈰三元復合氧化物;摩爾比;甲苯
0 前言
鑒于貴金屬過高的成本,將過渡金屬復合氧化物用于催化降解揮發性有機污染物(VOCs)被廣泛關注[1]。其中,銅錳二元復合氧化物常被用作VOCs催化燃燒的催化劑[2-4],但其起燃溫度與完全燃燒溫度仍較高[4-5]。人們發現稀土氧化物CeO2具有較好的儲氧功能,可促進晶格氧的移動性,降低催化劑的完全燃燒溫度[6-9]。因此,常通過氧化鈰與過渡金屬氧化物形成Cu-Ce[7-8]、Mn-Ce[7,9]、Ce-Zr[10]等體系的二元催化劑來提高催化劑的催化性能。Cai LN[11]研究了Fe、Zn和Ce元素的加入對銅錳催化劑活性的影響,結果顯示CuMnOx-Ce催化劑催化氧化CO的性能最佳。但不同加入量對銅錳催化劑的物相與性能的影響報道仍較少。本文將通過制備不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑,探討不同銅錳鈰摩爾比與催化劑的物相、比表面積與氧移動性的關系,進一步研究其對催化劑性能的影響。
1 實驗方法
1.1催化劑的制備方法
將15mL Cu(NO3)2溶液(c1mol/L)和15 mL Mn(NO3)2溶液(c2,mol/L)及10 mL Ce(NO3)3溶液(c3,mol/L)混合均勻后配成浸漬液,各硝酸鹽的濃度c見表1。將浸漬液加熱到50℃時,加入2.5000 g的TiO2(銳鈦礦:晶紅石摩爾比為4∶1,平均粒度:20 nm)粉末,控制催化劑的負載量為40%(以CuO,MnO2,CeO2的質量計算)。于50℃下磁力攪拌5 h,然后置于80℃水浴中旋轉蒸發除去水分,再在120℃下烘干12 h,得到Cu-Mn-Ce/TiO2-x-y-z催化劑(其中x、y和z分別代表Cu、Mn和Ce的摩爾比)。
將上述烘干的催化劑置于管式爐中,500℃焙燒4 h(焙燒氣氛為空氣,流速為10mL/min,升溫速率為2℃/min)。然后,催化劑經壓片、粉碎、篩分,取其中40~60目顆粒用于催化燃燒甲苯的實驗。
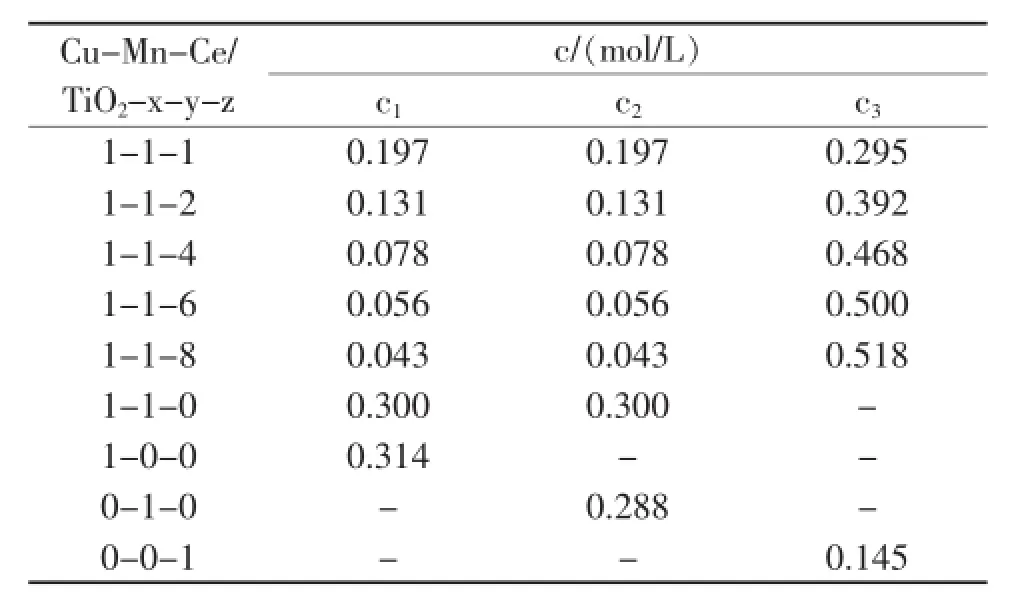
表1 浸漬液Cu(NO3)2、Mn(NO3)2和Ce(NO3)3的濃度Table 1.Concentration of impregnation solution of Cu(NO3)3、Mn(NO3)3and Ce(NO3)3
1.2催化劑表征
采用荷蘭PNAlytical公司的X'Pert PRO型的X射線衍射儀(XRD)對樣品的物相進行表征分析,得到催化劑的晶型和晶粒大小。分析條件為Cu Kα射線(波長為0.1541 nm),工作電壓40 kV,電流40 mA,掃描范圍2θ為10°~80°,掃描步進0.0167°/s,通過Scherrer公式計算D(hkl)。
氫氣程序升溫還原(H2-TPR)表征以體積分數為5%H2-95%Ar為還原氣體,尾氣用GC1690氣相色譜儀在線分析。當系統基線穩定后以10℃/min的速度從30℃升溫至810℃,得到樣品的H2-TPR圖譜用于分析催化劑中氧的移動性。
1.3催化評價條件
銅錳鈰三元復合氧化物催化劑催化燃燒性能的評價在常壓氣-固反應裝置上進行。催化劑填充在內徑為10 mm,長為500 mm的不銹鋼反應管的恒溫區,反應溫度范圍為150℃~400℃。催化劑(40~60目)用量為0.5 g(體積為0.6mL)。甲苯蒸汽是在10℃恒溫水浴中采用空氣鼓泡法得到,鼓泡氣流速是5 mL/min,并用另一路空氣進行稀釋,稀釋氣流速是175 mL/min,最終得到進氣濃度為1000 ppm的甲苯蒸汽加空氣的混合氣體。混合反應氣空速為18000 h-1。反應尾氣通過GC9650色譜儀進行在線分析。本實驗采用甲苯的轉化率(X)和CO2的選擇性(S)來表征催化劑的催化性能。

(式中Ain為進入催化劑之前甲苯的峰面積;Aout為經過催化劑后出口的甲苯的峰面積;[CO2]為反應得到CO2的組分濃度;[CO]為反應不完全燃燒產生CO的組分濃度;[CxHy]為不完全燃燒產生的CxHy的組分濃度;x為CxHy的碳原子數)
2 結果討論
2.1Cu/Mn/Ce摩爾比對晶相結構的影響
研究表明,在浸漬法制備負載型催化劑過程中,浸漬液濃度、浸漬時間、銅錳鈰摩爾比、銅錳鈰負載量、焙燒溫度都會影響催化劑的晶相結構。其中銅錳鈰的摩爾比對晶相結構和晶粒大小的影響較大。圖1和表2是不同銅錳鈰摩爾比的三元復合氧化物催化劑的XRD分析結果。
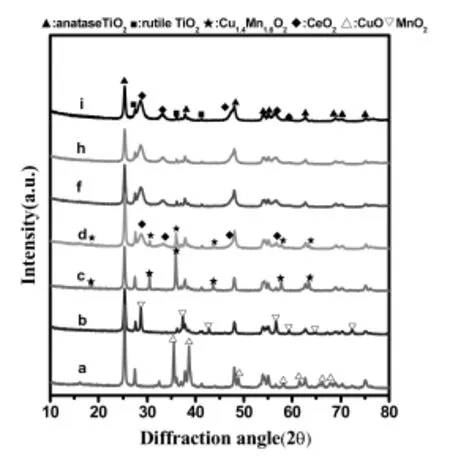
圖1 不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑的XRD圖Fig.1.XRD patterns of Cu-Mn-Ce ternary catalysts with different Cu/Mn/Cemolar ratios prepared by impregnationmethod
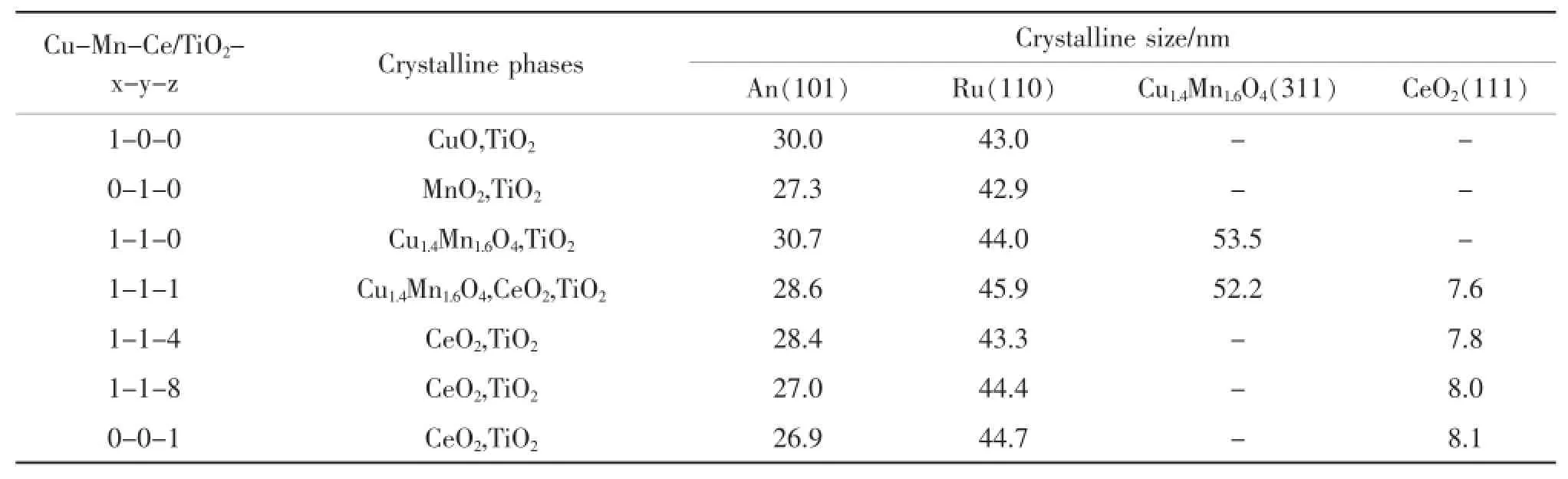
表2 不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑的活性組成和晶粒大小Table 2.Crystalline phases and crystalline size of Cu-Mn-Ce ternary catalystswith different Cu/Mn/Ce molar ratios prepared by impregnationmethod
圖1為不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑的XRD衍射圖。在圖1中,a,b和c分別是CuO,MnO2以及CuO和MnO2以1∶1的摩爾比負載在載體TiO2上的XRD圖。在a和b中,除了銳鈦礦型的二氧化鈦(TiO2-An)(PDF21-1272)和金紅石型的二氧化鈦(TiO2-Ru)(PDF21-1276)相態外,分別只有CuO(PDF48-1548)相態和MnO2(PDF24-0735)相態。當Cu/Mn的摩爾比為1:1時,在XRD圖c中CuO和MnO2的特征峰消失,代之以由氧化銅和氧化錳形成的新相Cu1.4Mn1.6O4(PDF 35-1030)的衍射峰。當鈰加入后,圖d中同樣沒有觀察到CuO和MnO2的特征峰,Cu1.4Mn1.6O4的衍射峰變弱,同時出現了新的CeO2(PDF43-1002)相態。隨著CeO2相對含量增加(Cu/Mn/Ce的摩爾比為1∶1∶4,1∶1∶8和0∶0∶1),其XRD圖(圖1中f,h,i)基本沒有變化,只觀察到CeO2的相態,不僅沒有觀察到氧化銅和氧化錳的特征峰,就是Cu1.4Mn1.6O4的衍射峰也消失了。可能原因是形成了粒度很小、分散度更高的、XRD無法檢測到的Cu1.4Mn1.6O4晶相[12]。也有可能是氧化銅和氧化錳以無定形的氧化態負載在載體TiO2表面,以致XRD無法檢測到。可以推知較高含量的CeO2可以高度分散Cu1.4Mn1.6O4,更高含量的CeO2甚至阻礙了Cu1.4Mn1.6O4的形成。
2.2Cu/Mn/Ce摩爾比對H2-TPR峰溫的影響
金屬氧化物的H2-TPR峰溫是催化劑的重要性質之一,代表了催化劑的氧化能力。一般而言,單一的金屬氧化物都有特定的H2-TPR峰溫,但當兩種或三種金屬氧化物形成三元催化劑時,會引起其中氧化物H2-TPR峰溫的變化。主要是由于催化劑各氧化物之間的相互作用影響了氧的移動性,從而影響了氧化物的被還原性[13]。為了研究不同銅錳鈰摩爾比的銅錳鈰三元復合氧化物催化劑的氧化能力,本實驗對其進行了H2-TPR表征。

圖2 不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑的H2-TPR圖Fig.2.H2-TPR profiles of Cu-Mn-Ce ternary catalystswith diffferent Cu/Mn/Cemolar ratios prepared by impregnationmethod
圖2為不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑的H2-TPR圖。從圖中可看出,不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑的H2-TPR峰型各不相同。當Cu/Mn/Ce摩爾比為1∶0∶0時,H2-TPR圖a中存在著溫度為212℃和343℃的兩個還原峰,它們分別對應的是無定形的CuO的還原峰和晶化程度較好的大顆粒CuO的還原峰[14],說明CuO在低溫段對氫有較強的氧化作用。當Cu/Mn/Ce摩爾比為0∶1∶0時,圖b中存在著溫度為469℃和523℃的兩個還原峰,根據XRD可知,這兩個峰是MnO2的還原峰[7],MnO2在中溫段對氫有較強的氧化作用。當Cu/Mn/Ce摩爾比為0∶0∶1時,圖i中存在著溫度為769℃的還原峰,這個峰是CeO2的還原峰[7,15],說明CeO2在高溫段對氫有較強的氧化作用。當催化劑Cu/Mn/Ce摩爾比為1∶1∶0時,H2-TPR圖c中存在著溫度為241℃和396℃的兩個還原峰,低溫峰為復合氧化物中銅的還原峰,高溫峰為復合氧化物中錳的還原峰[16],與圖a,b的單一氧化物的還原峰比,相應的峰溫都有所降低,說明形成復合氧化物有利于氧的移動。隨著CeO2的加入,三元復合氧化物催化劑的H2-TPR圖發生了變化。當銅錳鈰摩爾比為1∶1∶1時,對應的d圖中出現了5個峰,216℃對應于無定形的CuO還原峰,754℃對應于CeO2的還原峰,344℃為一定粒度的CuO還原峰,278℃為復合氧化物中銅的還原峰,416℃為復合氧化物中錳的還原峰,278℃和416℃這兩個峰由圖c的2個峰轉化而來,且都向高溫方向平移。可能原因是氧化鈰的作用。氧化鈰的加入對銅錳復合氧化物的氫還原峰起到兩個作用:一方面,復合氧化物中的銅、錳與CeO2之間存在著更強的相互作用,導致還原峰向高溫方向移動;另一方面,氧化鈰的加入可以高度分散Cu1.4Mn1.6O4,導致還原峰向低溫方向移動。結合XRD可知,CeO2的加入后Cu1.4Mn1.6O4晶粒略微變小(表2和圖1),復合氧化物中的銅、錳與CeO2之間存在著更強的相互作用,導致其還原峰向高溫方向移動。隨著CeO2進一步添加至銅錳鈰摩爾比為1∶1∶4時,圖f中出現4個還原峰,213℃對應于無定形的CuO還原峰,749℃對應于CeO2的還原峰,252℃與393℃為復合氧化物中銅、錳還原峰,此時形成的復合氧化物Cu1.4Mn1.6O4晶粒更小以至于XRD中未能出現衍射峰,此時氧化鈰的加入使Cu1.4Mn1.6O4高度分散,還原峰溫降低。但CeO2的過多加入阻礙了Cu1.4Mn1.6O4復合氧化物的形成。由此不難理解銅錳鈰摩爾比為1∶1∶8時,圖h中的5個還原峰向高溫方向移動,結合H2-TPR圖的a,b和i可知,215℃和305℃為CuO還原峰,434℃和466℃為MnO2的還原峰,750℃為CeO2的還原峰,還原峰分別接近單一的氧化銅,氧化錳和氧化鈰的還原峰。
2.3銅錳鈰三元復合催化劑的摩爾比對甲苯催化性能的影響
工業生產中所產生的揮發性氣體大多是有機污染物。在這些揮發性有機污染物(VOCs)中,尤以結構穩定的含苯環的系列物(如苯,甲苯等)居多,對人體危害大。為此,實驗中以甲苯催化燃燒作為探針反應,以考察不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合催化劑的催
化燃燒性能。實驗結果如圖3、4所示。
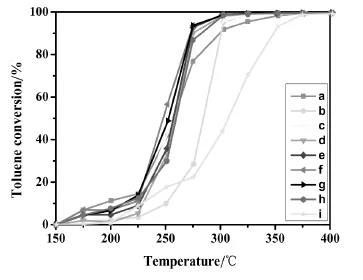
圖3 不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合催化劑催化氧化甲苯的轉化率與床層溫度的關系Fig.3.Toluene conversion as a function of the reaction temperature for Cu-Mn-Ce ternary catalystswith diffferent Cu/Mn/Cemolar ratios prepared by impregnationmethod
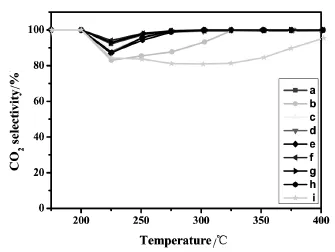
圖4 不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合催化劑催化氧化甲苯產生CO2的選擇性與床層溫度的關系Fig.4.CO2selectivity as a function of the reaction temperature for Cu-Mn-Ce ternary catalysts with diffferent Cu/Mn/Cemolar ratios prepared by impregnationmethod
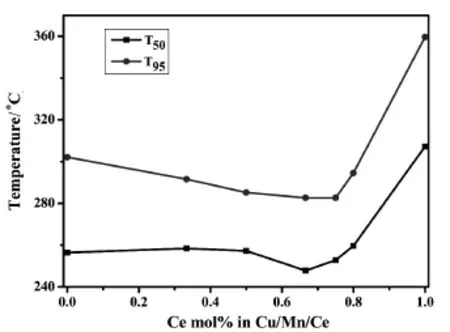
圖5 不同Cemol%的銅錳鈰三元復合氧化物催化劑的T50和T95Fig.5.T50,T95for Cu-Mn-Ce ternary catalystswith diffferent Cemol%prepared by impregnation method
由圖3可知,隨著反應溫度的升高,不同Cu/ Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑對甲苯的催化轉化率影響趨勢大致相同,分為三個階段:在低溫階段(150℃~225℃)時,甲苯的轉化率隨溫度呈緩慢增加的趨勢;當床層溫度達到225℃之后,反應劇烈,甲苯的轉化率曲線呈直線上升;直到反應溫度達到275℃后,轉化率趨于緩慢增加。具體分析,在不同反應溫度區間,不同的催化劑所表現的反應活性不同。在低溫階段,不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑對甲苯的轉化率相近。當床層溫度高于225℃時,可將不同Cu/Mn/Ce摩爾比的銅錳鈰催化劑分為三類情況來考察。第一類催化劑是負載有單金屬氧化物催化劑,即Cu/Mn/Ce摩爾比分別為1∶0∶0,0∶1∶0和0∶0∶1的CuO/TiO2、MnO2/TiO2和CeO2/TiO2催化劑。圖3和圖4的結果表明,無論是甲苯的轉化率,還是CO2的選擇性,其催化性能的順序是CuO/TiO2優于MnO2/TiO2,MnO2/TiO2優于CeO2/TiO2,與它們的H2-TPR峰溫從低到高的順序是一致的,這說明具有較低H2-TPR峰溫的氧化物催化劑有更好的催化氧化性能。第二類是二元復合氧化物催化劑Cu-Mn-Ce/TiO2-1-1-0(即Cu-Mn/TiO2催化劑)。從圖上可以看出,其甲苯的轉化率和CO2的選擇性明顯好于上述任一單金屬氧化物催化劑。由圖1可知,這時形成了復合氧化物新相Cu1.4Mn1.6O4,且有更低的氧化銅和氧化錳的H2-TPR峰溫(見圖2中c)。由此可以推知,復合氧化物Cu1.4Mn1.6O4是具有更高催化活性的新相。在新相Cu1.4Mn1.6O4中,Cu2+和Mn4+通過相互間的協同作用[17],各自都具有較單獨存在時更低的H2-TPR峰溫,從而有更高的催化活性。第三類是引進CeO2后的Cu/Mn/Ce三元復合氧化物催化劑(Cu-Mn-Ce/TiO2-1-1-1,Cu-Mn-Ce/ TiO2-1-1-2,Cu-Mn-Ce/TiO2-1-1-4,Cu-Mn-Ce/ TiO2-1-1-6和Cu-Mn-Ce/TiO2-1-1-8)。從圖5可以看出隨著CeO2的增加,催化劑的T95先減小后增加,催化劑Cu-Mn-Ce/TiO2-1-1-4(圖f)的T95最低(282.6℃),其甲苯的轉化率和CO2的選擇性最佳。結合圖1與圖2可知,當摻鈰量較少時,銅錳元素主要以較大顆粒的Cu1.4Mn1.6O4尖晶石形式分散在載體和CeO2上,復合氧化物中的銅、錳與CeO2之間存在著更強的相互作用,催化劑的還原峰溫較高,其T95仍較高;隨著鈰的進一步增加,復合物Cu1.4Mn1.6O4的晶粒度變小至XRD都無法觀察到衍射峰,此時高度分散的復合氧化物形成了更多的活性中心,更好地促進氧的流動,從而有更好的催化氧化甲苯的性能;但當鈰的含量增至1∶1∶8時,過多的Ce元素阻礙了Cu1.4Mn1.6O4的形成,此時沒有了Cu1.4Mn1.6O4活性中心的催化劑是銅、錳、鈰三種氧化物的簡單混合,氧的流動性減弱,氧化銅和氧化錳的H2-TPR峰溫升高,催化性能降低。
3 結論
本實驗采用浸漬法制備不同Cu/Mn/Ce摩爾比的銅錳鈰三元復合氧化物催化劑,對催化劑進行了XRD和H2-TPR表征分析,并對催化劑進行催化燃燒甲苯的性能評價。XRD和H2-TPR表征了不同Cu/Mn/Ce摩爾比對催化劑晶相和還原溫度的影響。結果表明:銅錳復合氧化物Cu1.4Mn1.6O4形成催化活性中心,一定量CeO2的加入可以高度分散活性中心Cu1.4Mn1.6O4,催化劑的還原峰溫降低;但更多量的CeO2會阻礙Cu1.4Mn1.6O4的形成,催化劑的還原峰向高溫移動。因此,當Cu/Mn/Ce摩爾比為1∶1∶4時,銅錳氧化物形成的活性中心Cu1.4Mn1.6O4高度分散在供氧中心CeO2與載體上,此時的還原峰溫度最低,氧移動性最強,催化劑的轉化率(T95為282.6℃)與CO2選擇性(100%)最高,催化氧化甲苯的性能最佳。
[1]Wang C H.Al2O3-supported transition-metal oxide catalysts for catalytic incineration incineration of toluene[J]. Cheosphere,2004,55:11-17.
[2]LiW B,Zhuang M.MCM-41 supported Cu-Mn catalysts for catalytic oxidation of toluene at low temperatures[J].J. Phys.Chem.B,2006,110:21568-21571.
[3]Chen H H,Zhang H P.Fabrication of porous copper/manganese binary oxides modified ZSM-5 membrane catalyst and potential application in the removal of VOCs[J]. Chemical Engineering Journal,2014,258:133-142.
[4]Van H V,Jamal B,A?ssa Ould-Dris,etal.Catalytic oxidation of volatile organic compounds on manganese and cop per oxides supported on titania[J].American Institute of Chemical Engineers Journal,2008,54(6):1585-1591
[5]Maria R M,Fabiola N A.Catalytic combustion of n-hexane over alumina supported Mn-Cu-Ce catalysts[J].CatalLett,2013,143:1003-1011.
[6]Larsson P O,Andersson A A.Oxides of copper,ceria promoted copper,manganese and coppermanganese on Al2O3for the combustion of CO,ethyl acetate and ethanol[J].Appl.Catal.B:environ,2000,24:175-192.
[7]Saleh M S,Dimitris IK,Xenophon EV,et al.Catalytic oxidation of toluene over binary mixtures of copper,man ganese and cerium oxides supported onγ-A l2O3[J].Applied Catalysis B:Environmental,2011,103:275-286.
[8]Unmesh M,Hilde P,Vitaliy B,et al.Nature of the active sites for the total oxidation of toluene by CuO-CeO2/Al2O3[J].Journal of Catalysis,2012,295:91-103.
[9]Dimitrios D,Theophilos I.VOC oxidation over MnOx-CeO2catalysts prepared by a combustion method[J].Applied Catalysis B:Environmental,2008,84:303-312.
[10]Beatriz de Rivas,Jose I.Gutiérrez-Ortiz,Rubén López-Fonseca,et al.Analysis of the simultaneous catalytic combustion of chlorinated aliphatic pollutants and toluene over ceria-zirconiamixed oxides[J].Applied Catalysis A:General,2006,314:54-63.
[11]Cai L N,Hu ZH,Peter B,et al.The effect of doping transition metal oxides on coppe manganese oxides forthe catalytic oxidation of CO[J].Chinese Journal of Catalysis,2014,35:159-167.
[12]Lu H F,Zhou Y,Han W F,ed al.Promoting effect of ZrO2carrier on activity and thermal stability of CeO2-based oxides catalysts for toluene combustion[J].Applied Catalysis A:General,2013,464:101-108.
[13]He C,Yu Y,Shen Q,et al.Catalytic behavior and synergistic effect of nanostructured mesoporous CuO-MnOx-CeO2catalysts for chlorobenzene destruction[J].Applied surface science,2014,297:59-69.
[14]MoralesM R,Barbero B P,Lopez T.Evaluation and characterization of Mn-Cu mixed oxide catalysts supported on TiO2and ZrO2for ethanol total oxidation[J].Fuel,2009,88: 2122-1229.
[15]Markaryan G L,Ikryannikova L N,Muravieva G P,et al. Red-ox properties and phase composition of CeO2-ZrO2and Y2O3-CeO2-ZrO2solid solutions[J].Colloids and SurfacesA:Physicochemical and Engineering Aspects,1999,151:435-447.
[16]MoralesM R,Barbero B P,Cadus L E.Total oxidation of ethanol and propane over Mn-Cumixed oxide catalysts[J]. App lied Catalysis B:Environmental,2006,67:229-236.
[17]Porta P,Moretti G,Musicanti M,et al.Characterization of copper-manganese mixed oxides[J].Catalysis Today,1991,9:211-218.
Study of the Cu/M n/Ce Ternary Catalysts on Performance for Catalytic Oxidation of Toluene
ZHU Hong-na1,LV De-yi1,HUAN Chang-yong2*
(1.College of Chemical Engineering,Zhejiang University of Technology,Hangzhou,Zhejiang 310014,China;2.National Post-Doctoral Scientific Research Center of Ningbo Petrochemical Economic& Technological Development Zone,Ningbo,Zhejiang 315204,China)
The different Cu/Mn/Cemolar ratio of Cu-Mn-Ce ternary catalystswere prepared by impregnation method and characterized by XRD and H2-TPR.Their performances for catalytic oxidation of toluene were evaluated in a fixed-bed tubular reactor,and the correlation of catalytic activity with active phases and oxygen mobility was discussed.The results showed that copper manganese oxide formed a new phase Cu1.4Mn1.6O4as the catalytic center,then the addition of CeO2highly dispersing Cu1.4Mn1.6O4increased the oxygenmobility of catalysts.So the Cu/Mn/Cemolar ratio 1∶1∶4 of Cu-Mn-Ce ternary catalyst showed the the best catalytic activity,with a lowest reaction temperature(T95was 282.6℃)and 100%selectivity of CO2.
copper-manganese-cerium ternary oxide catalyst;molar ratio;toluene
1006-4184(2016)10-0029-07
2016-04-08
朱紅娜(1991-),女,碩士研究生。E-mail:337089446@qq.com。
郇昌永,E-mail:huanmy2006@163.com。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
