HPLC法同時測定五倍子發酵百藥煎中沒食子酸及鞣花酸含量△
2016-09-25 06:47:02王瑞生張振凌王勝超李柯柯孫翼飛
中國現代中藥 2016年7期
關鍵詞:檢測
王瑞生,張振凌,王勝超,李柯柯,孫翼飛
(河南中醫學院,河南 鄭州 450008)
HPLC法同時測定五倍子發酵百藥煎中沒食子酸及鞣花酸含量△
王瑞生,張振凌*,王勝超,李柯柯,孫翼飛
(河南中醫學院,河南 鄭州450008)
目的:建立同時測定五倍子發酵百藥煎中抗氧化活性成分沒食子酸和鞣花酸含量的方法,并對不同批次五倍子發酵百藥煎中的含量進行比較。方法:反相高效液相色譜法,WatersSymmmetryShieldTMRP18(4.6×250mm,5μm)C18色譜柱;流動相:乙腈-0.1%三氟乙酸水,梯度洗脫;流速1.0mL·min-1;檢測波長254nm;柱溫35℃。結果:五倍子沒食子酸和鞣花酸含量分別為3.94%,0.25%;發酵成百藥煎含量增加,分別為14.20%,0.26%。結論:所建方法操作簡便、準確、重復性好,適用于同時測定五倍子發酵百藥煎中沒食子酸和鞣花酸的含量,含量測定結果為百藥煎的藥用研究和產品的開發提供了依據。
百藥煎;沒食子酸;鞣花酸;含量測定
百藥煎始載于《丹溪心法》[1],為五倍子同茶葉等經發酵制成的塊狀物。性味:酸甘,平。具有潤肺化痰,生津止渴的功效。臨床多用于治療久咳痰多,咽痛,便血,久痢脫肛,口瘡,牙疳,癰腫瘡瘍[2]。五倍子主要含沒食子酸和鞣質類成分,發酵工藝對其有效成分的含量具有一定的差別[3]。目前關于百藥煎的質量控制多數停留在性狀和沒食酸子酸含量測定[4],對于鞣花酸含量測定文獻報道不多。由于鞣花酸抗病毒、抗氧化、鎮靜等作用[5]與百藥煎抗炎、抗菌、鎮咳等藥效作用一致[6],將鞣花酸含量作為百藥煎質量控制的指標,具有重要的參考價值。因此,本文建立同時測定百藥煎藥材中沒食子酸和鞣花酸含量的HPLC方法,比較發酵對百藥煎中沒食子酸和鞣花酸的含量的影響,為百藥煎的質量控制和開發應用提供更全面的科學依據。
1 儀器與材料
1.1儀器
高效液相色譜分析儀(Waters,Waters2489UV檢測器),breeze數據處理系統,KQ-500DV型數控超聲波清洗器(昆山市超聲儀器有限公司),BS210S萬分之一電子天平(北京賽多利斯天平有限公司),BT25S十萬分之一電子天平(北京賽多利斯天平有限公司)。
1.2試劑
沒食子酸標準品(成都普斯生物科技有限公司,批號:P0380052,純度>98.5%),鞣花酸標準品(成都普斯生物科技有限公司,批號:P1440035,純度>98%),乙腈(迪馬科技有限公司),三氟乙酸(分析純),甲醇(天津四有精細化學品有限公司,批號:140325,一級色譜純),雙蒸水(自制),安琪釀酒曲(湖北宜昌安琪有限公司,批號:20150120),綠茶(河南信陽茶葉專業市場)。
1.3藥材
五倍子購于安徽亳州中藥飲片廠,批號:20130812,經河南中醫學院張振凌教授鑒定為漆樹科植物鹽膚木RhuschinensisMill.等樹上寄生倍蚜科昆蟲角倍蚜或倍蛋蚜后形成的蟲癭,符合《中國藥典》2010年版:“五倍子”項下的各項規定。百藥煎為本實驗室炮制,批號為20141203-z,20141219-z,20150123-z,20150129-z,20150731-z,及杭州桐君堂生物科技有限公司提供,批號為20150301-q。
2 方法與結果
2.1樣品的制備
2.1.1五倍子 將五倍子藥材洗凈,60℃干燥10h,粉碎,過80目篩。
2.1.2百藥煎 置60℃烘箱中烘干,粉碎,過四號篩,備用。
2.2供試品溶液的制備
分別取各批次五倍子及百藥煎粉末(過四號篩)約0.2g,精密稱定,加入甲醇50mL,稱定重量,放置2h,超聲提取40min,放至室溫,補足重量,過濾,取續濾液,用0.45μm微孔濾膜過濾,即得供試品溶液。
2.3對照品溶液的制備
精密稱取沒食子酸標準品0.02009g,置于25mL容量瓶,甲醇溶解定容,得濃度為0.8036mg·mL-1沒食子酸標準品溶液;精密稱取鞣花酸標準品0.00339g,置于100mL容量瓶,甲醇溶解定容,移取1mL,置于2mL容量瓶,甲醇定容,得0.01695mg·mL-1鞣花酸標準品溶液。
2.4色譜條件考察
色譜柱:WatersSymmmetryShieldTMRP18(4.6×250mm,5μm);其他條件進行研究如下。
2.4.1提取方法考察[7]考察了超聲提取和回流提取2種提取方法,依次進行HPLC分析,結果顯示超聲提取效率更高,選擇超聲為提取方法。選取50%甲醇,95%乙醇和甲醇3個提取溶劑進行了考察,結果顯示甲醇對百藥煎中沒食子酸及鞣花酸的提取率最高,因此選擇甲醇為提取溶劑。比較了不同的超聲時間(20,40,60min),結果表明超聲40min和60min提取率均較高,二者之間無顯著性差異,因此選擇超聲時間為40min。
2.4.2洗脫程序考察
2.4.2.1方法一[8]:流動相為乙腈和1%冰乙酸,梯度洗脫,流速1mL·min-1,柱溫35℃,檢測波長為248nm,進樣量10uL。梯度洗脫程序為0~10min,18%乙腈;10~15min,18%~20%乙腈。
2.4.2.2方法二[9]:流動相為乙腈(A)-0.25%甲酸溶液(B),梯度洗脫,流速0.85mL·min-1,柱溫25℃,檢測波長260nm,進樣量10uL。梯度洗脫程序為0~6min,3%~5%B;6~15min,5%~8%B;15~17min,8%~15%B;17~25min,15~18%B;25~33min,18%~22%B;33~35min,22%~25%B;35~40min,25%~50%B;40~45min,50%~100%B。
2.4.2.3方法三[10]:流動相為甲醇-0.1%磷酸溶液,梯度洗脫,流速1.0mL·min-1,柱溫為30℃,檢測波長280nm,進樣量10uL。梯度洗脫程序為0~15min,10%甲醇;15~20min,10%~50%甲醇;20~40min,50%甲醇。
2.4.2.4方法四:流動相為乙腈-1%三氟乙酸,梯度洗脫,流速1mL·min-1,柱溫35℃,檢測波長為254nm[11],進樣量10uL。梯度洗脫程序為0~10min,18%乙腈;10~15min:18%~20%乙腈。
對比以上四種方法得到的色譜圖,根據沒食子酸及鞣花酸色譜峰的峰型,峰面積,分離度及峰高,選擇方法四檢測百藥煎中沒食子酸及鞣花酸含量測定的色譜條件。
2.4.3流動相考察 對比乙腈-0.1%甲酸,乙腈-0.1磷酸,乙腈-0.1%冰醋酸,乙腈-0.1%三氟乙酸,根據色譜圖峰面積、分離度及峰型,選擇乙腈-0.1%三氟乙酸為流動相,結果見圖1。

A:乙腈-0.1%甲酸,B:乙腈-0.1%磷酸,C:乙腈-0.1%冰醋酸,D:乙腈-0.1%三氟乙酸圖1 百藥煎不同流動相色譜圖
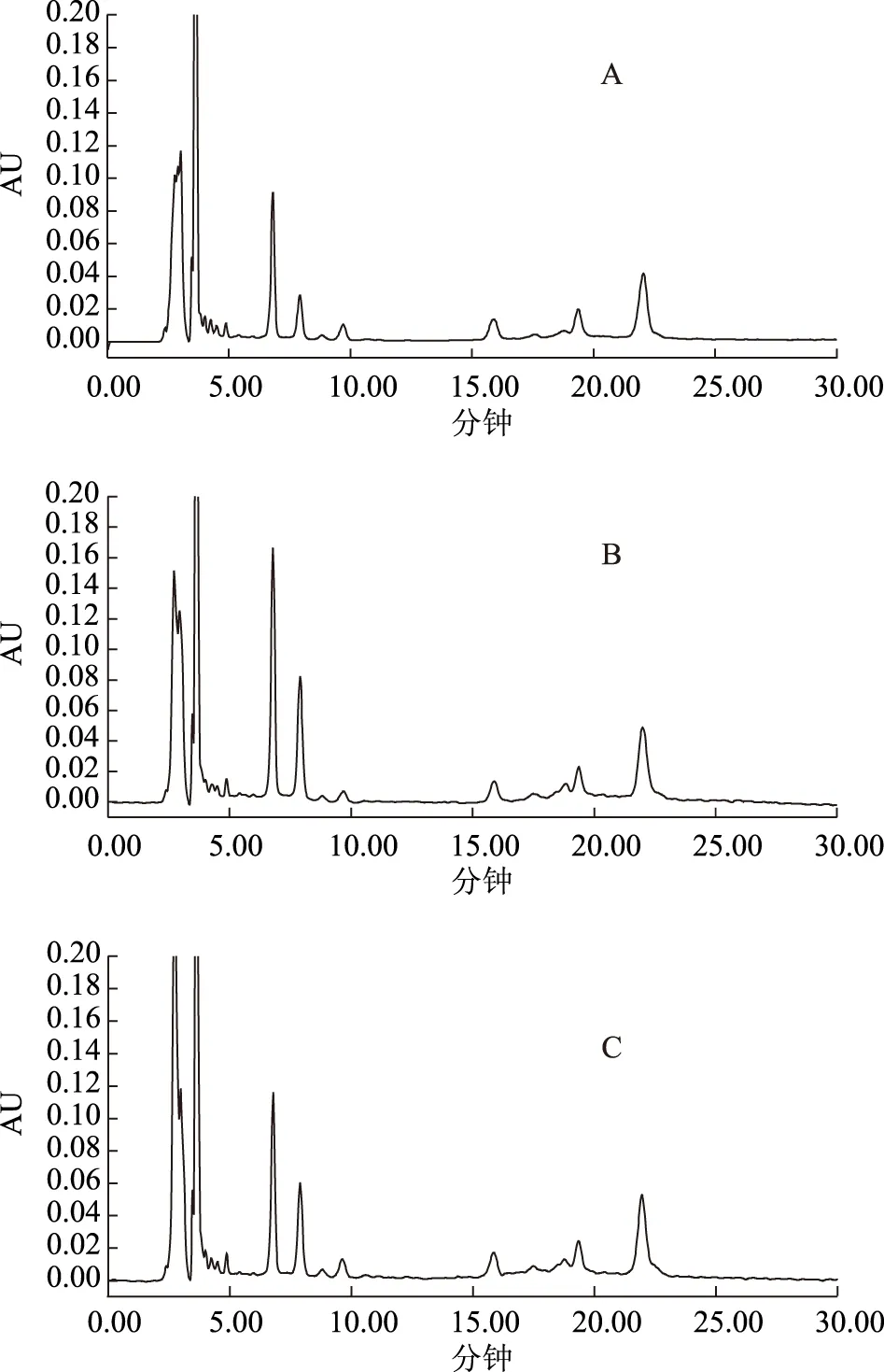
2.4.4檢測波長考察 將沒食子酸標品,鞣花酸標品及百藥煎樣品進行全波長掃描,掃描范圍:190nm-600nm,掃描間距:1nm,結果見圖2。沒食子酸在246nm,254nm,286nm,293nm處有最大吸收;鞣花酸在254nm,365nm處有最大吸收;百藥煎樣品在238nm,250nm,262nm,286nm,305nm處有最大吸收。綜合實驗數據及參考文獻選擇對比248nm,254nm,260nm,280nm四個波長下色譜圖,最終選擇254nm為檢測波長,結果見圖3。
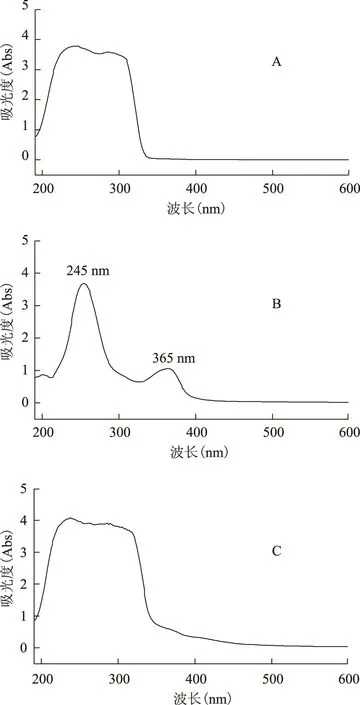
A:沒食子酸標品圖,B:鞣花酸標品圖,C:百藥煎樣品圖圖2 百藥煎標準品及樣品全波長掃描圖
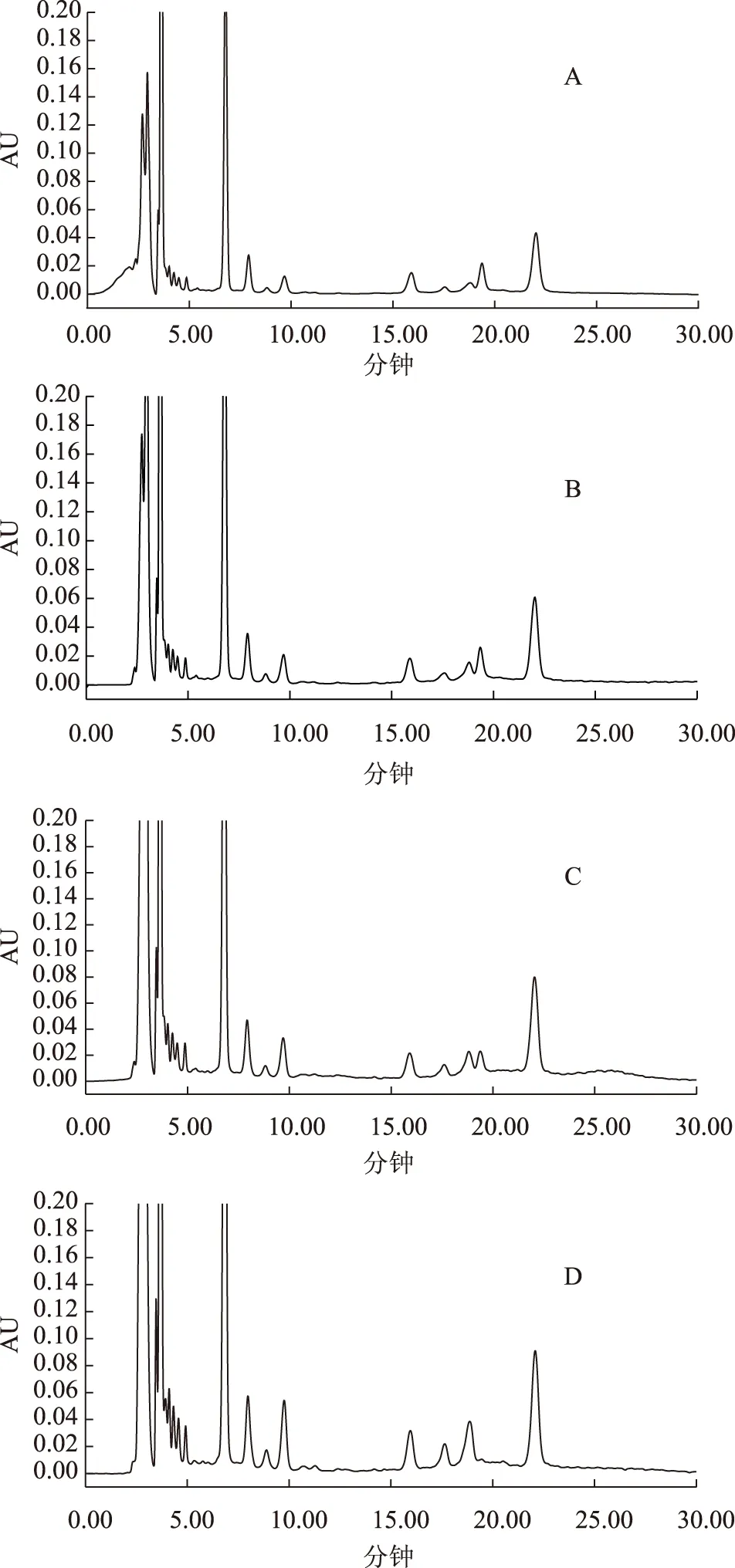
2.4.5柱溫考察 對比30℃,35℃,40℃三個檢測溫度下的色譜圖,30℃色譜圖峰高和分離度均較低,35℃與40℃色譜圖差別較小,考慮到40℃,溫度較高對儀器及色譜柱有影響,選擇35℃為檢測柱溫,結果見圖4。
經以上比較,確定流動相:乙腈-0.1%三氟乙酸水,按表1梯度洗脫;流速1mL·min-1;檢測波長254nm;柱溫35℃;進樣量10μL。

表1 百藥煎沒食子酸及鞣花酸含量測定梯度洗脫程序
A:248 nm,B:254 nm,C:260 nm,D:280 nm圖3 百藥煎不同檢測波長色譜圖
A:30 ℃,B:35 ℃,C:40 ℃圖4 百藥煎不同檢測柱溫色譜圖
2.5方法學考察
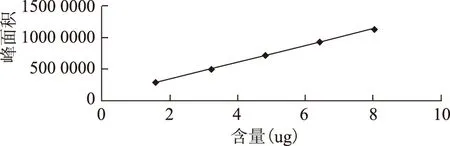
圖5 沒食子酸標準曲線
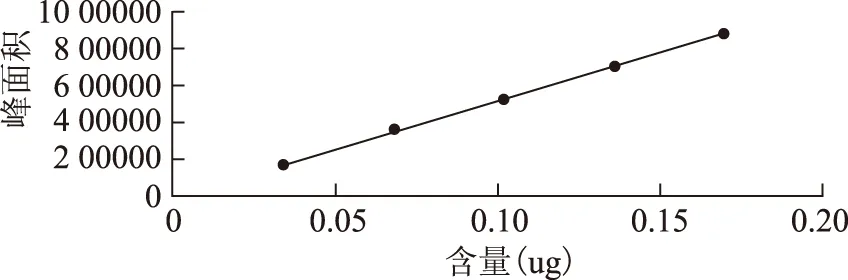
圖6 鞣花酸標準曲線
2.5.1線性關系考察 取對照品溶液,分別進樣2、4、6、8、10uL,以峰面積為縱坐標,進樣量(ug)為橫坐標,繪制標準曲線,Y沒食子酸=1337951.72X+613105(R2=0.9997),Y鞣花酸=5240147.49X-1416(0.9994)。結果表明沒食子酸和鞣花酸分別在1.6072~8.0360ug,0.0339~0.1695ug呈良好線性,見圖5,圖6。2.5.2精密度實驗 取20150129百藥煎粉末(過四號篩)約0.2g,精密稱定,按2.3.項下供試品溶液的制備方法操作,進樣量10μL,連續進樣5次,記錄沒食子酸、鞣花酸峰面積,以沒食子酸、鞣花酸峰面積計算,測定值的RSD分別為3.41%,1.11%,表明系統的精密度較好。
2.5.3 穩定性試驗 取20150129百藥煎粉末(過四號篩)約0.2g,精密稱定,按2.3.項下供試品溶液的制備方法操作,分別在0h、2h、4h、8h、12h檢測,記錄沒食子酸、鞣花酸峰面積,以沒食子酸、鞣花酸峰面積計算,測定值的RSD分別為 0.28%,0.33%,表明樣品在12h內穩定性良好。
2.5.4 重復性試驗 取20150129百藥煎粉末(過四號篩)5份,每份約0.2g,精密稱定,按2.3.項下供試品溶液的制備方法操作,分別進樣10μL,記錄沒食子酸、鞣花酸峰面積,以沒食子酸、鞣花酸峰面積計算,測定值的RSD分別為 1.09%,2.02%,表明方法重復性良好。
2.5.5 加樣回收率試驗 取已知含量的百藥煎樣品(20150129)粉末5份,加入一定量的沒食子酸和鞣花酸對照品,測定并計算回收率,結果見表2,表3。

表2 沒食子酸加樣回收率(n=6)
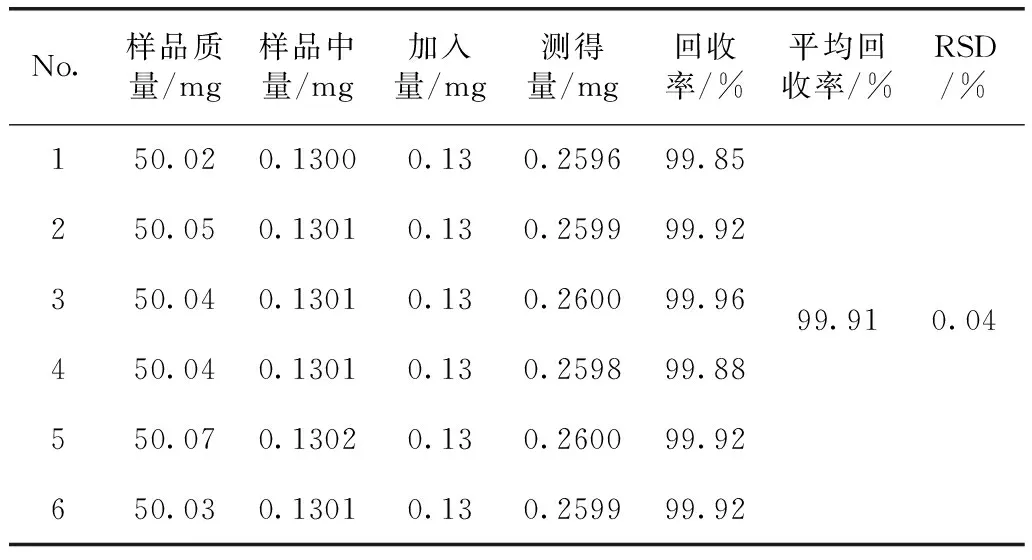
表3 鞣花酸加樣回收率(n=6)
2.6 含量測定 稱取3份五倍子樣品和6批次百藥煎樣品,按2.3項下制備下列批次的待測樣品溶液,進樣量為10 μL,分別注入高效液相色譜儀,測定色譜峰峰面積,對照品溶液和百藥煎供試品的HPLC色譜圖,見圖7。結果見表4。
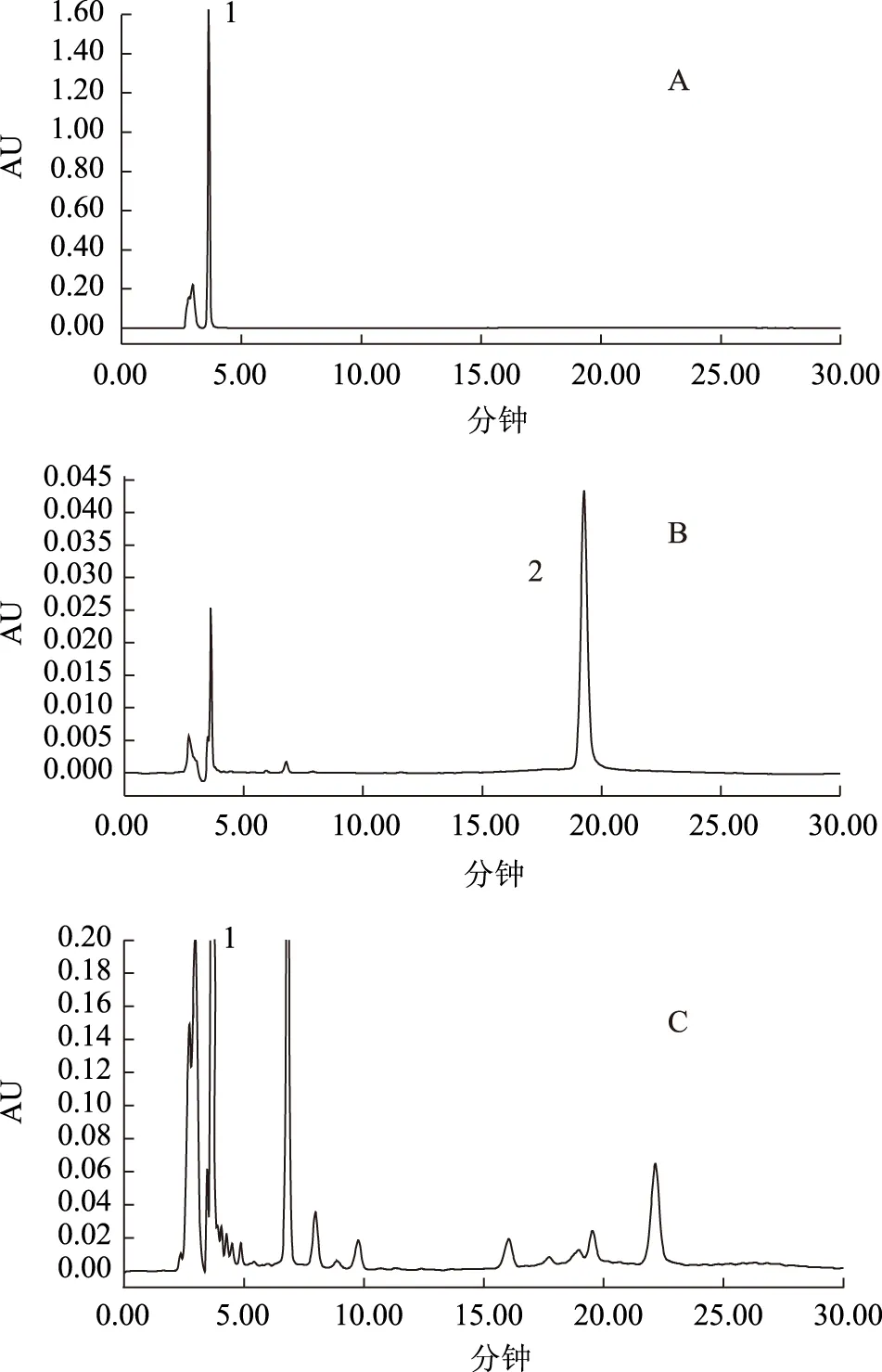
1:沒食子酸 2:鞣花酸;圖A:沒食子酸標品圖,B:鞣花酸標品圖,C:百藥煎樣品圖圖7 百藥煎標準品及樣品HPLC
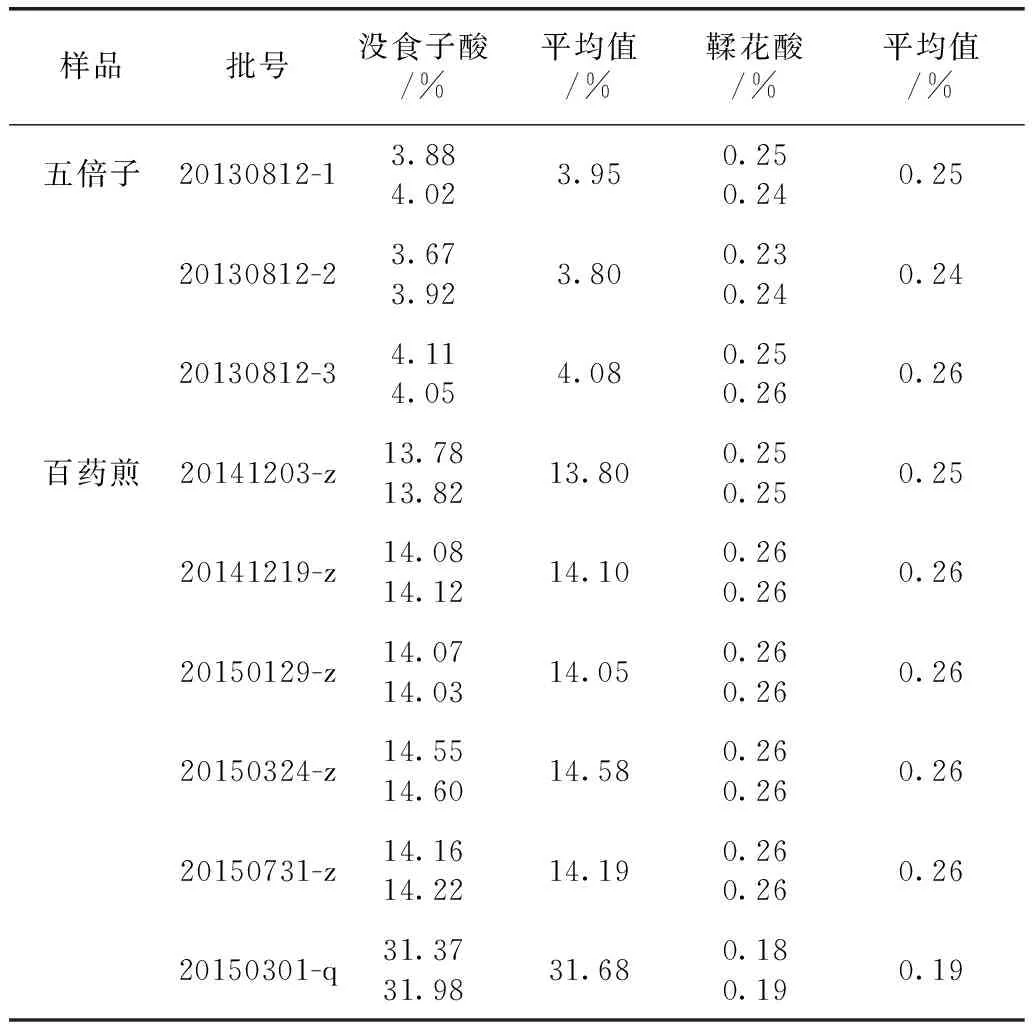
樣品批號沒食子酸/%平均值/%鞣花酸/%平均值/%五倍子20130812-13.884.023.950.250.240.2520130812-23.673.923.800.230.240.2420130812-34.114.054.080.250.260.26百藥煎20141203-z13.7813.8213.800.250.250.2520141219-z14.0814.1214.100.260.260.2620150129-z14.0714.0314.050.260.260.2620150324-z14.5514.6014.580.260.260.2620150731-z14.1614.2214.190.260.260.2620150301-q31.3731.9831.680.180.190.19
3 討論
3.1
本文建立同時測定百藥煎中沒食子酸及鞣花酸含量的方法,比較了不同提取方法工藝條件以及不同檢測波長、不同檢測柱溫、不同梯度洗脫比例對沒食子酸及鞣花酸含量的影響。與文獻報道[8]的同時測定五倍子中沒食子酸及鞣花酸含量的方法對比,本文所建立方法同時測定五倍子發酵前后沒食子酸及鞣花酸含量,比較發酵前后成分含量變化,對于五倍子發酵過程的控制具有一定指導意義。五倍子中鞣質含量高達60%~80%[12],發酵后鞣質的水解產物包括沒食子酸和鞣花酸。本次測定結果證明五倍子沒食子酸含量為3.94%,實驗室發酵炮制百藥煎含量為14.20%,增加3.6倍;五倍子鞣花酸含量為0.25%,發酵制成百藥煎后含量為0.26%,含量稍有增加。考慮五倍子發酵炮制百藥煎的過程中加入了茶葉、茶汁及釀酒曲,質量增加40%,因此,計算得沒食子酸含量實際增加5.04倍,鞣花酸含量實際增加量為0.11%。說明發酵過程中五倍子鞣質水解,部分生成沒食子酸及鞣花酸。
3.2
有文獻報道,鞣花酸具有抗癌、抗突變性能,對人體免疫缺陷病毒的抑制作用抗氧化作用,具有凝血功能[5],對多種細菌、病毒都有很好的抑制作用[13],有降壓、鎮靜作用[14]。本研究對生產企業提供的百藥煎進行研究,發現與本實驗室制備的百藥煎不同,沒食子酸含量增加8倍,鞣花酸含量降低0.07%,說明發酵時間、菌種、發酵程度的控制等工藝條件對五倍子發酵炮制的百藥煎成分的種類與含量均有顯著影響。因此,五倍子發酵炮制百藥煎的轉化是將鞣質全部或者盡可能多地轉化成為沒食子酸,還是保留一部分中間成分,尚需要進一步通過藥理功效甚至是中藥臨床功效的進一步驗證研究。其處方、工藝條件尤其是發酵程度的控制也需要進一步研究。
[1] 元·朱震亨著.丹溪心法.上海科學技術出版社,1963:96.
[2] 江蘇新醫學院編.中藥大辭典[Z],上海人民出版社,1977:865.
[3] 禹玉洪,韓小敏,張建麗,等.百藥煎發酵炮制的酒曲篩選研究[C].第十七屆全國色譜學術報告會及儀器展覽會會議論文集,2009:1039-1040.
[4] 禹玉洪,韓小敏,張建麗,等.百藥煎發酵過程中關鍵菌株的確定[C].全國中藥創新與研究論壇·論文集,2009:123-127.
[5] 李素琴,袁其朋,徐健梅.鞣花酸的生理功能及工藝開發研究現狀[J].天然產物研究與開發,2001,13(5):71.
[6] 胡昌江,楊斂芳,瞿燕.改良百藥煎的主要藥效學研究[J].陜西中醫學院學報,2001,24(3):44-45.
[7] 彭璐,龔千鋒,李小寧,等.RP-HPLC測定五倍子3個炮制品中沒食子酸和鞣花酸的含量[J].中國實驗方劑學雜志,2015,21(18):76-79.
[8] 盛達成,陳凌,王文茂,等.高效液相色譜法同時分析五倍子提取物中的沒食子酸和鞣花酸[J].中國農學通報,2013,29(04):148-154.
[9] 丁楠,高曉黎.HPLC法測定石榴皮提取物中鞣花酸的含量[J].新疆醫科大學學報,2012,35(6):770-772.
[10] 張目,朱少娟,嚴澤民,等.HPLC測定地榆中沒食子酸和鞣花酸的含量[J].現代科學儀器,2009,5:69-70.
[11] 張海偉,張藝,楊繼家,等.HPLC測定不同批次藏藥三果湯中沒食子酸和鞣花酸的含量[J].中國實驗方劑學雜志,2013,19(10):95-97.
[12] 喬彩云,李建科.五倍子及五倍子單寧的研究進展[J].食品工業科技,2011,32(7):458-462.
[13] Haramo Tet al.Chem.Pharm.Bull.,1988,36(6).
[14] Gordon M H,and Jing An.J.Agric.Food Chem.,1995,43:1784-1788.
DeterminationofGallicAcidandEllagicAcidinGallnutFermentedChineseGallLeavenbyHPLC
WANGRuisheng,ZHANGZhenling*,WANGShengchao,LIKeke,SUNYifei
(HenanUniversityofChineseMedicine,Zhengzhou450008,China)
Objective:To determine the gallic acid and ellagic acid in the gallnut fermented Chinese gall leaven by HPLC and compare the content of different batches.Methods:The separation was performed on the Waters Symmmetry ShieldTMRP18(4.6×250mm,5μm) C18column,and acetonitrile-0.1% trifluoroacetic acid aqueous in gradient as mobile phase,the flow rate was1.0mL·min-1,and the detecting wavelength was set at254nm,the column temperature was kept at35℃.Results:The average content of gallic acid and ellagic acid in Chinese gall were3.94% and0.25%,respectively,and in Chinese gall leaven were14.20%,0.26%,respectively.Conclusion:The established method is suitable for the determination of gallic acid and ellagicacid in the gallnut fermented Chinese gall leaven.The results provide a basis for medicinal value research and product development of gallnut fermented Chinese gall leaven.
Chinese gall leaven;gallic acid;ellagic acid;determination
10.13313/j.issn.1673-4890.2016.7.007
2016-01-30)
六神曲等7種中藥發酵技術及規范化應用研究--百藥煎(201507004-03);呼吸疾病診療與新藥研發河南省協同創新中心-研究生科研創新基金項目(20140013)
*
張振凌,博士生導師;研究反向:中藥飲片及新藥研究;Tel:(0371)65680970,E-mail:zhangzl6758@163.com
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
