治療性低溫對缺氧期心肌細胞自噬的影響及機制
2016-09-06 00:46:08楊羽菲趙叢楊平珍王先寶鄧意陳愛華
山東醫藥 2016年22期
楊羽菲,趙叢,楊平珍,王先寶,鄧意,陳愛華
(南方醫科大學珠江醫院,廣州510280)
?
·論著·
治療性低溫對缺氧期心肌細胞自噬的影響及機制
楊羽菲,趙叢,楊平珍,王先寶,鄧意,陳愛華
(南方醫科大學珠江醫院,廣州510280)
目的觀察治療性低溫(TH)在缺氧期對心肌細胞自噬活性及自噬流的影響,并探討其作用機制。方法取大鼠心肌細胞株H9c2,分為對照組、缺氧組、TH干預組、雷帕霉素干預組、雷帕霉素+TH干預組,對照組置于37 ℃培養箱中培養,不作處理;缺氧組和TH干預組構建缺氧模型后,分別置于37、32 ℃培養箱中培養;雷帕霉素干預組和雷帕霉素+TH干預組于構建模型前2 h加入雷帕霉素0.1 μmol/L,構建模型后分別置于37、32 ℃培養箱中培養。采用Western blot法檢測對照組、缺氧組和TH干預組自噬相關蛋白自噬微管相關蛋白輕鏈3B(LC3B)、p62、Beclin-1蛋白表達,以及應用雷帕霉素前后哺乳動物雷帕霉素靶蛋白(mTOR)、核糖體蛋白S6(S6)的表達差異;采用腺病毒GFP-mRFP-LC3熒光瞬時轉染技術檢測自噬流。結果①對照組、缺氧組和TH干預組LC3B、p62、Beclin-1蛋白表達:缺氧組和TH干預組LC3B、Beclin-1高于對照組(P均<0.05),缺氧組p62降低(P<0.05)。TH干預組LC3B明顯低于缺氧組,p62增高(P均<0.05)。缺氧組與TH干預組Beclin-1無統計學差異。②雷帕霉素處理后自噬蛋白LC3B、p62及mTOR通路相關蛋白p-mTOR、p-S6表達:TH干預組與缺氧組比較LC3B降低,而p62、p-mTOR和p-S6均升高(P均<0.05);雷帕霉素干預組與缺氧組比較LC3B升高,p62降低(P均<0.05),而p-mTOR和p-S6差異無統計學意義;雷帕霉素+TH干預組與TH干預組比較LC3B升高,p62、p-mTOR和p-S6均降低,結果均有統計學差異(P均<0.05);雷帕霉素+TH干預組與雷帕霉素干預組比較LC3B降低,p62升高(P均<0.05),而p-mTOR和p-S6差異無統計學意義。③雙熒光自噬流:缺氧組和TH干預組的黃點和紅點數多于對照組(P均<0.05),TH干預組的黃點與紅點數少于缺氧組(P均<0.05)。結論 TH能夠抑制缺氧期心肌細胞自噬,其機制可能與促進mTOR信號通路相關蛋白表達有關。
治療性低溫;心肌細胞;缺氧;自噬;雷帕霉素
治療性低溫(TH)能夠減少活性氧族(ROS)產生,改善細胞內離子水平,保持細胞內pH平衡[1~3],影響細胞能量代謝,減輕細胞損害,對缺氧缺血的心肌細胞有一定的保護作用,是臨床治療缺血性疾病的有效方法。自噬是指細胞質內大分子物質被包裹并傳遞至溶酶體進行降解,維持細胞內的物質與能量平衡。研究認為,自噬是心肌細胞存活與否的關鍵調節因素之一。雷帕霉素靶蛋白(mTOR)是在哺乳動物細胞中負向調控自噬的關鍵蛋白,其活化對自噬具有抑制作用[4]。mTOR通路是缺血期調控自噬的兩條經典通路之一。磷酸化的mTOR(p-mTOR)是mTOR通路活化的標志。核糖體蛋白6(S6)是mTOR下游的一個效應蛋白,磷酸化S6(p-S6)被認為是mTOR激活程度的重要標記蛋白。2014年1月~2016年1月,我們觀察了TH在大鼠心肌細胞缺氧期對H9c2細胞自噬活性相關蛋白[自噬微管相關蛋白輕鏈3B(LC3B)、p62及Beclin-1]表達的影響,并采用雷帕霉素抑制mTOR信號通路,觀察心肌細胞自噬活性相關蛋白和mTOR通路相關蛋白p-mTOR、p-S6的變化,探討TH治療心肌細胞缺氧損傷的作用機制。
1 材料與方法
1.1材料大鼠心肌細胞株H9c2購自美國ATCC公司。自噬蛋白LC3B、p62、Beclin-1相關一抗、mTOR信號通路相關一抗購于美國CST公司,相關二抗購于武漢博士德公司。雷帕霉素購于美國Sigma-Aldrich公司。
1.2心肌細胞自噬活性檢測采用Western blot法。將大鼠心肌細胞置于含10%胎牛血清的高糖DMEM培養基中,5% CO2、37 ℃培養箱中培養。細胞融合至70%~90%后用于實驗。將細胞分為對照組、缺氧組、TH干預組,對照組置于37 ℃培養箱中培養,不作處理;其余兩組向細胞中加入PBS,移入細胞缺氧盒,注入含5% CO2和95% N2的混合氣體構建缺氧模型,分別置于37、32 ℃培養箱中培養。向細胞懸液中加入蛋白裂解液,收集上清,加入15% SDS-PAGE分離蛋白,將凝膠蛋白轉至PVDF膜上,室溫封閉2 h,加入相應的一抗、二抗,4 ℃過夜孵育。以ECL試劑為底物,X線曝光,顯色、定影。采用Quantity One 4.6軟件對發光條帶的灰度值進行分析,以β-actin作為內參,計算LC3B、p62、Beclin-1蛋白的相對表達量。實驗重復3次。
1.3雷帕霉素對心肌細胞自噬及mTOR通路蛋白的影響觀察將細胞分為缺氧組、TH干預組、雷帕霉素干預組、雷帕霉素+TH干預組。缺氧組、TH干預組處理同1.2,雷帕霉素干預組與雷帕霉素+TH干預組于缺氧造模前2 h加入雷帕霉素0.1 μmol/L,造模后分別置37、32 ℃培養箱中培養。采用Western blot法檢測LC3B、p62及p-mTOR和p-S6蛋白表達,加入相應抗體,計算其相對表達量,操作及計算方法參照1.2。
1.4心肌細胞自噬流檢測采用GFP-mRFP-LC3熒光瞬時轉染技術。用腺病毒GFP-mRFP-LC3瞬時轉染心肌細胞,培養36 h后分為對照組、缺氧組、TH干預組,分別處理3 h。由于GFP 綠色熒光和mRFP 紅色熒光在自噬體同時存在,在紅、綠熒光合成圖像中呈黃色;但在自噬溶酶體酸性環境中GFP 綠色熒光降解,只剩下紅色熒光。以合成圖像中的黃色亮點(自噬體)和紅色亮點(自噬溶酶體)數量反映細胞內自噬囊泡的數量。分組處理細胞后在400倍熒光顯微鏡下觀察,隨機采集300 個細胞內的紅、綠色LC3 熒光圖,計數其合成圖像中平均每個細胞含有的黃色亮點和紅色亮點的個數。實驗重復3次。
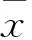
2 結果
2.1各組LC3B、p62、Beclin-1蛋白相對表達量比較缺氧組和TH干預組LC3B、Beclin-1高于對照組(P均<0.05),缺氧組p62降低(P<0.05)。TH干預組LC3B低于缺氧組,p62增高(P均<0.05)。缺氧組與TH干預組Beclin-1無統計學差異。見表1。
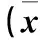
表1 各組LC3B、p62和Beclin-1蛋白相對表達量比較±s)
注:與對照組比較,*P<0.05;與缺氧組比較,#P<0.05。
2.2雷帕霉素處理后各組LC3B、p62、p-mTOR、p-S6蛋白相對表達量比較TH干預組與缺氧組比較LC3B降低,而p62、p-mTOR和p-S6均升高(P均<0.05);雷帕霉素干預組與缺氧組比較LC3B升高,p62降低(P均<0.05),而p-mTOR和p-S6差異無統計學意義;雷帕霉素+TH干預組與TH干預組比較LC3B升高,p62、p-mTOR和p-S6均降低,結果均有統計學差異(P均<0.05);雷帕霉素+TH干預組與雷帕霉素干預組比較LC3B降低,p62升高(P均<0.05),而p-mTOR和p-S6差異無統計學意義。見表2。
2.3各組細胞雙熒光自噬流檢測結果缺氧組和TH干預組的黃點和紅點多于對照組(P均<0.05),TH干預組的黃點與紅點少于缺氧組(P均<0.05)。見表3。
3 討論
細胞自噬是指在細胞內通過形成雙層脂質膜包裹細胞內細胞器或細胞內物質(自噬小體),隨之與溶酶體結合并降解的過程[5]。LC3B是自噬體膜上的標記蛋白,細胞內LC3B蛋白的變化通常與自噬活性聯系密切,被廣泛用于自噬活性的研究[6]。另一個被廣泛應用的自噬標記蛋白p62,其降解程度能有效反映自噬活性[7]。Beclin-1是細胞自噬的一個關鍵調控因子,是自噬體形成不可或缺的條件[8]。研究顯示,TH是一種多重機制的治療方式,它對細胞的保護機制多與應用TH的開始時間有關。缺血期應用TH要優于再灌注期,不僅可以減少缺血期產生的病理產物ROS等,還可能影響再灌注期間炎癥信號通路等[9]。盡早應用TH可使機體獲益更多。TH能有效抑制缺血期H9c2細胞的壞死與凋亡[10],缺血前或再灌注治療前應用TH均能減少H9c2細胞凋亡。然而TH調控自噬及其相關機制少見報道。
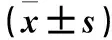
表2 各組LC3B、p62、p-mTOR、p-S6蛋白相對表達量比較
注:與缺氧組比較,*P<0.05;與TH干預組比較,#P<0.05;與雷帕霉素干預組比較,△P<0.05。

表3 各組熒光點數比較(個,±s)
注:與對照組比較,*P<0.05;與缺氧組比較,#P<0.05。
近期文獻報道,在大鼠心肌頓抑模型中,TH能有效恢復神經功能,并通過抑制自噬改善海馬旁回腦細胞活性[11]。另外,TH可以通過抑制凋亡、降低過度自噬修復缺血損傷的腦脊髓神經功能[12,13]。在缺血再灌注損傷的心肌細胞中,TH改善細胞活性并下調自噬水平[14]。本研究表明,TH可降低缺氧期間H9c2心肌細胞自噬水平(降低LC3B、升高p62),在缺氧損傷環境下表現出抑制細胞自噬活性。Beclin-1在自噬誘導的起始階段起重要作用[15],但缺氧組與TH干預組相比,Beclin-1表達并無統計學差異。表明TH對自噬的影響并不依賴Beclin-1的表達,與以往報道[14]相符。
為了探討TH對自噬活性的調節機制,我們進一步對mTOR信號通路進行研究。mTOR信號通路的激活對細胞自噬活性具有負性調節作用[16]。作為mTOR信號通路的關鍵成員,mTOR和S6蛋白磷酸化與自噬的調控密切相關[17]。p-mTOR及p-S6表達可反映mTOR通路激活程度[18],表達升高提示mTOR信號通路高度激活,并會對自噬活性造成顯著抑制[18]。而雷帕霉素作為自噬活性的經典激活劑,可抑制mTOR信號通路,從而激活自噬。本研究顯示,TH能通過促進mTOR和S6蛋白磷酸化來激活mTOR通路(使p-mTOR、p-S6表達升高),并抑制缺氧期間H9c2的自噬水平(使LC3B降低、p62升高)。然而當雷帕霉素作用于TH干預下的缺氧期H9c2細胞時,TH對自噬的抑制作用完全消失,取而代之的是自噬活性的強烈激活,并且雷帕霉素也逆轉了TH對p-mTOR和p-S6的促表達作用。簡而言之,雷帕霉素的介入抑制了mTOR信號通路(p-mTOR、p-S6表達降低),同時也強烈激活了原本受到TH抑制的自噬活性,說明TH或可通過激活mTOR信號通路抑制自噬活性。
自噬流是指自噬的完整過程,自噬小體的雙層膜結構包裹部分胞質和細胞內需降解的細胞器、蛋白質等形成自噬體,最后與溶酶體融合形成自噬溶酶體,降解其所包裹的內容物,以實現細胞穩態和細胞器的更新[19]。因此,較之以LC3B和p62蛋白表達來反映細胞自噬活性,自噬流的檢測更加直觀,可以更方便地對自噬形成過程的各個階段進行分析解讀。本研究顯示,與對照組相比,缺氧組與TH處理組的黃色熒光點數、紅色熒光點數增多,提示缺氧促進了H9c2心肌細胞自噬小體和自噬溶酶體的升高,而TH則部分減少了缺氧后H9c2心肌細胞自噬小體和自噬溶酶體,與缺氧組相比,TH處理組的自噬體、自噬溶酶體減少。說明TH能夠通過抑制缺氧期H9c2心肌細胞自噬小體的形成,從而抑制自噬流水平。
綜上所述,TH可通過mTOR相關信號通路對細胞自噬流進行調控,抑制缺氧期H9c2心肌細胞的自噬流。此研究揭示了TH在心血管系統中又一新的機制,為缺血性心臟病的治療提供了理論依據。
[1] Aoki M, Nomura F, Stromski ME, et al. Effects of ph on brain energetics after hypothermic circulatory arrest[J]. Ann Thorac Surg, 1993,55(5):1093-1103.
[2] Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia[J]. Crit Care Med, 2009,37(7 Suppl):S186-202.
[3] Shao ZH, Sharp WW, Wojcik KR, et al. Therapeutic hypothermia cardioprotection via akt- and nitric oxide-mediated attenuation of mitochondrial oxidants[J]. Am J Physiol Heart Circ Physiol, 2010,298(6):2164-2173.
[4] Pattingre S, Espert L, Biard-Piechaczyk M, et al. Regulation of macroautophagy by mtor and beclin 1 complexes[J]. Biochimie, 2008,90(2):313-323.
[5] Pyo JO, Nah J, Jung YK. Molecules and their functions in autophagy[J]. Exp Mol Med, 2012,44(2):73-80.
[6] Kabeya Y, Mizushima N, Ueno T, et al. Lc3, a mammalian homologue of yeast apg8p, is localized in autophagosome membranes after processing[J]. EMBO J, 2000,19(21):5720-5728.
[7] Pankiv S, Clausen TH, Lamark T, et al. P62/sqstm1 binds directly to atg8/lc3 to facilitate degradation of ubiquitinated protein aggregates by autophagy[J]. J Biol Chem, 2007,282(33):24131-24145.
[8] Funderburk SF, Wang QJ, Yue Z. The beclin 1-vps34 complex--at the crossroads of autophagy and beyond[J]. Trends Cell Biol, 2010,20(6):355-362.
[9] Lampe JW, Becker LB. State of the art in therapeutic hypothermia[J]. Annu Rev Med, 2011,62(1):79-93.
[10] Lin CH, Wu WS, Lin MT, et al. Attenuating ischemia-induced h9c2 myoblasts apoptosis by therapeutic hypothermia[J]. Am J Med Sci, 2010,339(3):258-265.
[11] Lu J, Qian HY, Liu LJ, et al. Mild hypothermia alleviates excessive autophagy and mitophagy in a rat model of asphyxial cardiac arrest[J]. Neurol Sci, 2014,35(11):1691-1699.
[12] Seo JY, Kim YH, Kim JW, et al. Effects of therapeutic hypothermia on apoptosis and autophagy following spinal cord injury in rats[J]. Spine, 2015,40(12):883.
[13] Choi KE, Hall CL, Sun JM, et al. A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice[J]. FASEB J, 2012,26(7):2799-2810.
[14] Cheng BC, Huang HS, Chao CM, et al. Hypothermia may attenuate ischemia/reperfusion-induced cardiomyocyte death by reducing autophagy[J]. Int J Cardiol, 2013,168(3):2064-2069.
[15] Kihara A, Noda T, Ishihara N, et al. Two distinct vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase y sorting in saccharomyces cerevisiae[J]. J Cell Biol, 2001,152(3):519-530.
[16] Dibble CC, Manning BD. Signal integration by mtorc1 coordinates nutrient input with biosynthetic output[J]. Nat Cell Biol, 2013,15(6):555-564.
[17] Tan VP, Miyamoto S. Nutrient-sensing mTORC: Integration of metabolic and autophagic signals[J]. J Mol Cell Cardiol, 2016.PMID:26773603
[18] Jung CH, Ro SH, Cao J, et al. Mtor regulation of autophagy[J]. FEBS Lett, 2010,584(7):1287-1295.
[19] Gatica D, Chiong M, Lavandero S, et al. Molecular mechanisms of autophagy in the cardiovascular system[J]. Circ Res, 2015,116(3):456-467.
Effect and mechanism of therapeutic hypothermia on autophagy in hypoxia cardiomyocytes
YANGYufei,ZHAOCong,YANGPingzhen,WANGXianbao,DENGYi,CHENAihua
(ZhujiangHospital,SouthernMedicalUniversity,Guangzhou510280,China)
ObjectiveTo investigate the effects of therapeutic hypothermia (TH) on autophagy and autophagic flux of cardiomyocytes in ischemia and to explore the underlying mechanism. MethodsH9c2 rat cardiomyocytes were divided into the control group, hypoxia group, hypoxia + TH group, rapamycin + hypoxia group and rapamycin + hypoxia+ TH group. Cells in the control group were normally cultured at 37 ℃ and received no further treatments. Cells with or without TH treatment in the other four groups were subjected to hypoxia in a hypoxic chamber at 32 ℃ or 37 ℃ respectively. Then, 0.1 μmol/L rapamycin was added 2 h before cells were subjected to hypoxia at 32 ℃ or 37 ℃. Western blotting was performed to detect protein expression of LC3B, p62, Beclin-1, phospho-mTOR (p-mTOR) and phospho-S6 (p-S6). The autophagic flux was detected under adenovirus GFP-mRFP-LC3 confocal laser scanning microscopy.Results①The protein expression of LC3B, p62 and Beclin-1 in the control group, hypoxia group and hypoxia + TH group: compared with the control group, LC3B and Beclin-1 expression was significantly increased in the hypoxia group and hypoxia + TH group (allP<0.05), while p62 expression was significantly decreased in the hypoxia group (P<0.05). Hypoxia + TH group exhibited lower LC3B expression as well as higher p62 expression as compared with that of the hypoxia group (P<0.05). There was no significant difference in Beclin-1 expression between the hypoxia group and hypoxia+TH group. ② The expression of autophagy-related proteins LC3B, p62 and mTOR pathway-related proteins p-mTOR and p-S6 after rapamycin treatment: compared with the hypoxia group, hypoxia + TH group showed decreased expression of LC3B and increased expression of p62, p-mTOR and p-S6 (allP<0.05). Compared with the hypoxia group, rapamycin+hypoxia group showed higher expression of LC3B and lower expression of p62 (allP<0.05), while there were no significant differences in p-mTOR and p-S6. Rapymycin + hypoxia+TH group showed increased LC3B and decreased p62, p-mTOR and p-S6 expression as compared with that of the hypoxia+TH group, and significant difference was found between them (allP<0.05). The rapamycin + hypoxia+TH group showed lower LC3B and higher p62 expression (allP<0.05), but there were no significant differences in p-mTOR and p-S6 expression between rapamycin + hypoxia+TH group and rapamycin +hypoxia group. ③ Detection of autophagic flux: compared with the control group, the green and red puncta were significantly higher in the hypoxia group and hypoxia + TH group (allP<0.05). Hypoxia + TH group showed less green and red puncta than those of the hypoxia group (allP<0.05). ConclusionTH inhibits the autophagic flux of hypoxic cardiomyocytes and the mechanism may be related to the activation of mTOR pathway-related proteins.
therapeutic hypothermia; cardiomyocytes; hypoxia; autophagy; rapamycin
國家自然科學基金資助項目(81400190,81270218);廣東省自然科學基金資助項目(2015A030310478);廣東省科技計劃項目(2014A020212191)。
楊羽菲(1989-),女,在讀碩士研究生,主要研究方向為自噬在心肌缺血損傷中的影響及機制。E-mail: lvyyang0608@163.com
簡介:陳愛華(1956-),男,博士,教授,主要研究方向為自噬在心肌缺血和缺血再灌注損傷中的影響及機制。E-mail: zj_chenaihua@126.com
10.3969/j.issn.1002-266X.2016.22.001
R541.2
A
1002-266X(2016)22-0001-04
2016-02-25)
