反相高效液相色譜法測定消渴平片中黃芪甲苷的含量Δ
2016-08-15 00:47:44周穎儀劉瀟瀟
中國醫(yī)院用藥評價與分析 2016年7期
周穎儀,劉瀟瀟
(廣東省藥品檢驗所中成藥室,廣東 廣州 510180)
?
反相高效液相色譜法測定消渴平片中黃芪甲苷的含量Δ
周穎儀*,劉瀟瀟#
(廣東省藥品檢驗所中成藥室,廣東 廣州510180)
目的:建立消渴平片中黃芪甲苷含量的測定方法。方法:采用薄層色譜法對制劑中黃連、葛根進行定性鑒別。采用反相高效液相色譜法測定制劑中黃芪甲苷的含量:色譜柱為Thermo BDS Hypersil C18(4.6 mm×250 mm,5 μm),流動相為乙腈-水(V∶V=32 ∶68),流速為1 ml/min,柱溫為30 ℃,檢測采用蒸發(fā)光散射檢測器。結果:定性鑒別專屬性強,陰性無干擾;黃芪甲苷質量范圍在0.196 2~10.172 0 μg之間與峰面積呈良好的線性關系,r=0.999 8,平均回收率為100.7%,RSD為2.1%(n=9)。結論:本研究結果準確、重復性好,能有效控制該制劑的質量。
消渴平片; 反相高效液相色譜法; 黃芪甲苷
消渴平片由黃芪、黃連、葛根等12味中藥組成,具有益氣養(yǎng)陰、清熱瀉火之效,臨床用于陰虛燥熱、氣陰兩虛所致的消渴病,癥見口渴喜飲、多食、多尿、消瘦、氣短、乏力、手足心熱,以及2型糖尿病見上述癥候者。消渴平片為《中華人民共和國藥典》(2015年版)任務品種,該品種原質量標準收載于《中華人民共和國藥典:一部》(2010年版)第1 042頁。本研究根據(jù)《2015年版藥典科研立任務單》的要求,采用薄層色譜法對消渴平片中黃連、葛根進行定性鑒別,使用反相高效液相色譜法對制劑中黃芪的黃芪甲苷進行定量測定,綜合評價該制劑的質量,為該制劑提供簡便、可靠的質量控制方法。
1 材料
1.1儀器
Agilent 1100型高效液相色譜儀;Alltech ELSD 2000ES型蒸發(fā)光檢測器;Sartorius-CP225D、Sartorius-CP224S電子天平;色譜柱:Thermo BDS Hypersil C18(4.6 mm×250 mm,5 μm),Merck Purospher?STAR RP-18(4.6 mm×250 mm,5 μm),資生堂C18 ACR C18(6 mm×250 mm,5 μm)。預制硅膠G薄層板,購于Merck公司、青島海洋化工廠分廠及煙臺市化學工業(yè)研究所。
1.2藥品與試劑
消渴平片(廣州中一藥業(yè)有限公司,批號:R00001、R00002、R00003;康普藥業(yè)股份有限公司,批號:20121109、20110809、20110523;吉林萬通藥業(yè)集團梅河藥業(yè)股份有限公司,批號:20110901、20111201;長春英平藥業(yè)有限公司,批號:20120901);黃芪甲苷對照品(批號:110781-200613)、黃連對照藥材(批號:913-200910)、鹽酸小檗堿對照品(批號:110713-200910)、葛根(甘葛藤)對照藥材(批號: 1175-200001)、葛根素(批號:110752-200912)均購于中國食品藥品檢定研究院,陰性樣品為本實驗室自制。乙腈為色譜純,水為純化水,其余試劑級別均為分析純。
2 方法與結果
2.1定性鑒別
2.1.1黃連的薄層鑒別:取本品2片,研細,加氨水2 ml潤濕,再用三氯甲烷液20 ml,超聲處理30 min,放冷,濾過,蒸干濾液,殘渣加1.5 ml甲醇使之溶解,作為供試品溶液。另取黃連對照藥材0.25 g,加25 ml甲醇,超聲處理(350 W,35 Hz)30 min,濾過,取續(xù)濾液,作為對照藥材溶液[1]。再取鹽酸小檗堿對照品適量,加甲醇溶解使成每1 ml含0.5 mg的溶液,作為對照品溶液。取缺黃連的陰性樣品,按供試品溶液的制備方法同法制成缺黃連陰性樣品溶液。吸取供試品溶液、對照藥材溶液、對照品溶液、缺黃連陰性樣品溶液各1 μl,分別點于同一硅膠G薄層板上,以甲苯-乙酸乙酯-甲醇-異丙醇-水 (V∶V∶V∶V∶V=6.0 ∶3.0 ∶2.0 ∶1.5 ∶0.3)為展開劑,置于用濃氨溶液預飽和20 min的展開缸里,展開,取出,晾干,在紫外光(365 nm)下檢視。供試品色譜中,在與對照藥材色譜和對照品色譜相應的位置上,顯相同顏色的熒光斑點,見圖1。
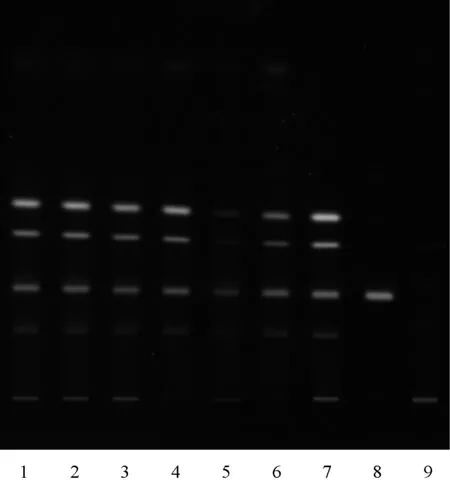
1~6.供試品; 7.黃連對照藥材; 8.對照品; 9.陰性樣品1-6.Test sample; 7.Coptidis Rhizoma reference herb; 8.Reference substance; 9.Negative sample圖1 黃連薄層色譜圖Fig 1 TLC chromatogram of Coptidis Rhizoma
2.1.2葛根的薄層鑒別:取本品20片,研細,加熱水適量使其溶解,放冷,加硅藻土適量,加乙酸乙酯100 ml,超聲30 min,放冷,濾過,濾液蒸干,殘渣加2 ml甲醇使其溶解,作為供試品溶液[2]。另取葛根對照藥材0.5 g,加20 ml甲醇,放置2 h,濾過,蒸干濾液,殘渣加甲醇1.5 ml使其溶解,取續(xù)濾液,作為對照藥材溶液。再取葛根素對照品,加甲醇溶解,制成每1 ml含1 mg的溶液,作為對照品溶液。取缺葛根的陰性樣品,按供試品溶液的制備方法同法制成缺葛根陰性樣品溶液。吸取供試品溶液、對照藥材溶液、對照品溶液、缺葛根陰性樣品溶液各2 μl,分別點于同一硅膠G薄層板上,以三氯甲烷-甲醇-水(V∶V∶V=7.00 ∶2.50 ∶0.25)為展開劑[3],展開,取出,晾干,在紫外光(365 nm)下檢視。供試品色譜中,在與對照藥材色譜和對照品色譜相應的位置上,顯相同顏色的熒光斑點,見圖2。
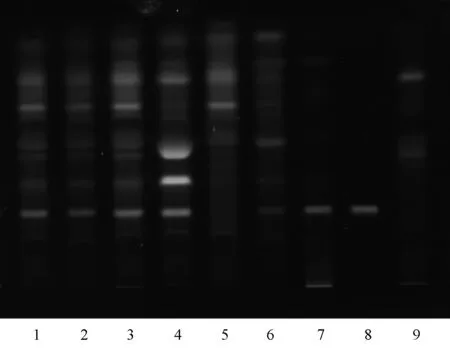
1~6.供試品; 7.葛根對照藥材; 8.對照品; 9.陰性樣品1-6.Test sample; 7.Puerariae lobatae radix reference herb;8.Reference substance; 9.Negative sample圖2 葛根薄層色譜圖Fig 2 TLC chromatogram of Puerariae Lobatae Radix
2.2含量測定
2.2.1色譜條件:色譜柱為Thermo BDS Hypersil C18(4.6 mm×250 mm,5 μm);以乙腈-水(V∶V=32 ∶68)[3]為流動相;流速為1 ml/min;柱溫為30 ℃;蒸發(fā)光散射檢測器檢測;氮氣流量為2.5 L/min;漂移管溫度為105 ℃時的色譜條件對黃芪甲苷的分離較好,靈敏度高,分析時間短。
2.2.2對照品溶液的制備:取黃芪甲苷對照品適量,精密稱定,加甲醇制成每1 ml含0.2 mg的溶液,即得。
2.2.3供試品溶液的制備:取本品,研細,取約6 g,精密稱定,置于具塞錐形瓶中,加50 ml甲醇,加熱回流45 min,放冷,過濾,用少量甲醇分次洗滌錐形瓶及濾紙,濾液和洗液合并,蒸干,殘渣加水20 ml使溶解,用水飽和的正丁醇振搖提取4次,每次40 ml,合并正丁醇提取液液,用氨試液洗滌2次,每次40 ml,棄去氨液,正丁醇液蒸干,殘渣加甲醇溶解,轉移至10 ml容量瓶中,加甲醇稀釋至刻度,搖勻,即得。
2.2.4缺黃芪陰性樣品溶液的制備:按供試品溶液的制備方法制成缺黃芪陰性樣品溶液。
2.2.5專屬性考察:吸取對照品溶液、供試品溶液與缺黃芪陰性樣品溶液各10 μl,注入高效液相色譜儀,結果顯示,缺黃芪陰性樣品無干擾,表明專屬性良好,見圖3。
2.2.6線性關系考察:分別配制0.019 62、0.058 86、0.196 20、0.392 40、0.508 60、1.017 20 mg/ml黃芪甲苷系列對照品溶液,設定進樣體積為10 μl,以峰面積的對數(shù)(Y)為縱坐標,質量的對數(shù)(X) 為橫坐標進行線性回歸,得黃芪甲苷的回歸方程為:Y=1.543 7X+7.223 5,r=0.999 8。結果表明,黃芪甲苷質量范圍在0.196 2~10.172 0 μg之間與峰面積呈良好的線性關系。
2.2.7精密度(重復性試驗):取本品(批號為R0001),研細,取約6 g,共取6份,精密稱定,按“2.2.3”項下方法制備供試品溶液,按上述色譜條件測定,6次操作之間的RSD為4.5%(n=6),說明本方法重現(xiàn)性良好。
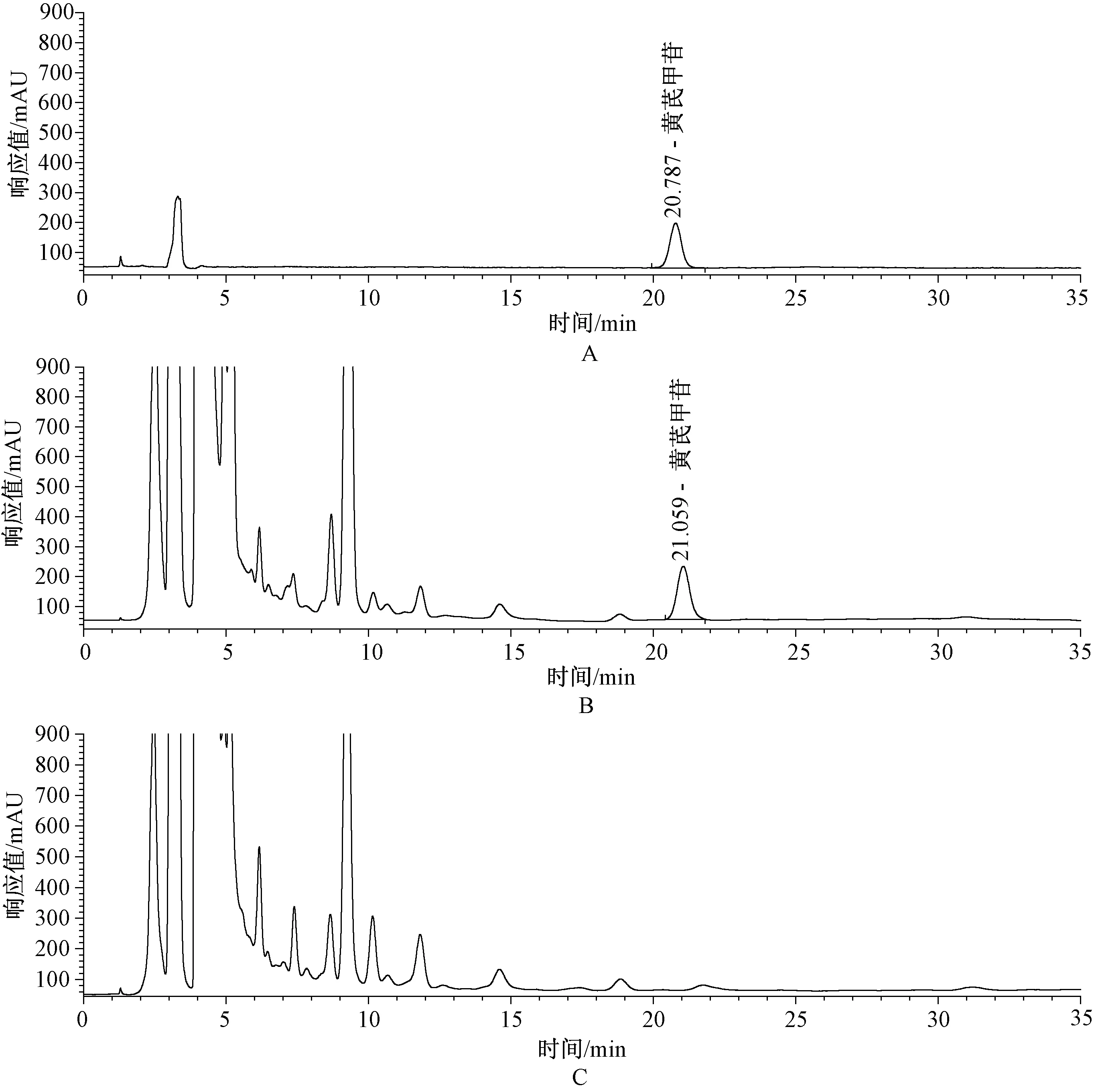
A.對照品; B.供試品; C.缺黃芪陰性樣品A.Reference substance; B.Test sample; C.Negative sample without Astragali Radix圖3 高效液相色譜圖Fig 3 HPLC chromatogram
2.2.8準確度(加樣回收率試驗):取本品(批號為R0001,含黃 芪甲苷0.436 7 mg/g)9份,每份3 g(含黃芪甲苷1.310 10 mg),分別按相當于該批號消渴平片中黃芪甲苷含量約80%(1.048 08 mg)、100%(1.310 10 mg)、120%(1.572 12 mg)各3份加入黃芪甲苷對照品,按含量測定方法測定,計算黃芪甲苷的回收率。結果顯示,平均回收率為100.7%,RSD為2.1%(n=9),說明方法的準確度較好。
2.2.9樣品含量測定:取來自4個企業(yè)的9批消渴平片,按“2.2.3”項下方法制備供試品溶液,按上述色譜條件測定,計算結果見表1。
3 討論
3.1黃連薄層鑒別
以黃連對照藥材和鹽酸小檗堿為對照,制訂黃連的薄層鑒別方法。分別考察了4種展開劑:(1)環(huán)己烷-乙酸乙酯-異丙醇-甲醇-水-三乙胺(V∶V∶V∶V∶V∶V=3.0 ∶3.5 ∶1.0 ∶1.5 ∶0.5 ∶1.0)[1];(2)乙酸乙酯-丙酮-甲酸-水 (V∶V∶V∶V=10 ∶7 ∶7 ∶1)[4];(3)正丁醇-冰醋酸-水 (V∶V∶V=7 ∶1 ∶2)[5];(4)甲苯-乙酸乙酯-甲醇-異丙醇-水 (V∶V∶V∶V∶V=6.0 ∶3.0 ∶2.0 ∶1.5 ∶0.3)[6]。結果表明,展開劑(1)斑點不清晰,Rf值偏低;展開劑(2)斑點Rf值偏高;展開劑(3)斑點清晰,Rf值偏適中;展開劑(4)斑點清晰, Rf值偏適中,且信息量優(yōu)于展開劑(3),因此,選擇展開劑(4)作為該薄層色譜的展開劑。
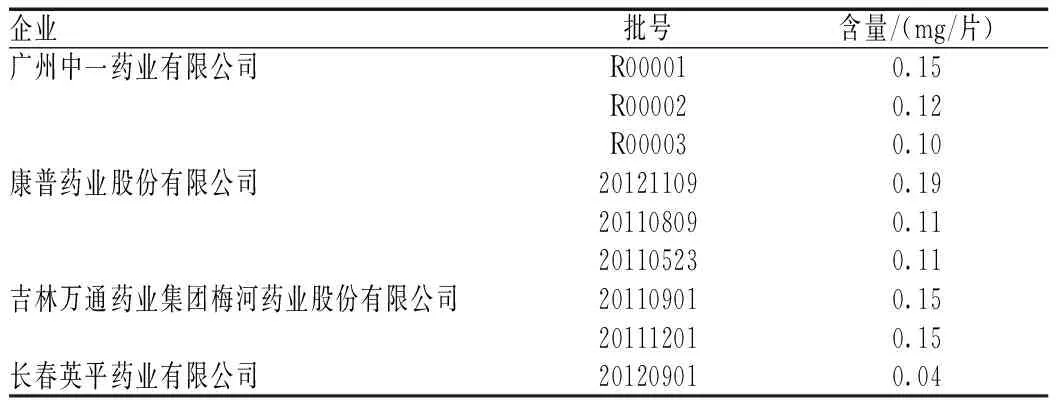
表1 樣品測定結果(n=9)Tab 1 Determination results of samples (n=9)
3.2黃芪甲苷的含量測定
根據(jù)《中華人民共和國藥典》(2010年版)黃芪藥材中黃芪甲苷的含量測定方法,進一步優(yōu)化,采用反相高效液相色譜法測定制劑中黃芪甲苷的含量。對于提取方法,考察了索氏提取(4 h)、超聲處理(1 h)、加熱回流提取(1 h)3種方法[7-9],結果表明,加熱回流提取1 h所得的黃芪甲苷含量較高,確定提取方法為加熱回流。對于提取時間,考察了加熱回流15、30、45、60 min,結果表明,加熱回流45 min所提取的黃芪甲苷含量較高,確定加熱回流提取時間為45 min。還考察了是否通過預處理好的D101大孔樹脂[10-12],結果表明,通過D101大孔樹脂與不通過D101大孔樹脂的供試品溶液測得的黃芪甲苷含量相近,從簡便操作考慮,選擇不通過D101大孔樹脂的提取方法。本品為薄膜衣片,考慮到除去包衣會增加檢驗步驟,并會造成誤差,故對是否除去包衣進行考察,結果表明,樣品除去與不除去包衣所測得黃芪甲苷的含量差異不大,為方便操作,采用不去包衣取樣。
3.3不同來源樣品的差異
觀察不同來源樣品所測得結果,可以發(fā)現(xiàn)部分供試品色譜中,在與對照藥材色譜和對照品色譜相應的位置上,雖然顯示相同顏色的熒光斑點,但其斑點較為模糊,說明含量較低。另外在黃芪甲苷的含量測定中,不同來源的黃芪甲苷含量存在一定的差異,其中長春英平藥業(yè)有限公司的消渴平片含量最低,與其他來源的差別較大。
綜上所述,本研究在《中華人民共和國藥典》(2010年版)消渴平片標準的基礎上,加以改進[13-15]。其中,薄層色譜法鑒別黃連、葛根操作方便、專屬性強,反相高效液相色譜法測定黃芪甲苷的含量結果準確,方法簡便、穩(wěn)定、專屬性強、重復性好,可以用于消渴平片的質量控制。
[1]國家藥典委員會.中華人民共和國藥典:2010年版:第一增補本[S].北京:中國醫(yī)藥科技出版社,2012:115,283-284,312-313.
[2]申桂霞.玉液消渴沖劑中黃芪葛根的薄層鑒別[J].中醫(yī)藥導報,2007,13(7):92.
[3]國家藥典委員會.中華人民共和國藥典:一部[S].2010年版.北京:化學工業(yè)出版社,2010:283-284,312-313.
[4]周紅超.薄層色譜法鑒別芩連胃康丸[J].中國醫(yī)藥導報,2011,8(4):55.
[5]黃有霖,潘馨.成方中黃連、黃柏的薄層鑒別的研究[J]海峽藥學,2003,15(2):36-37
[6]國家藥典委員會.中藥材薄層色譜彩色圖集[M].北京:人民衛(wèi)生出版社,2009:698.
[7]白娟,張潔,李成網(wǎng).HPLC-ELSD測定玉屏風軟膠囊中黃芪甲苷的含量[J].中國實驗方劑學雜志,2012,18(16):102-104.
[8]江延輝.HPLC-ELSD法測定復方芪參片中黃芪甲苷的含量[J].中醫(yī)現(xiàn)代中藥,2013,15(4):314-316.
[9]郭強.HPLC-ELSD法測定老年咳喘片中黃芪甲苷的含量[J].中醫(yī)研究,2013,26(6):76-78.
[10]李矗,劉圣,王珍.HPLC-ELSD法測定復方阿膠黃芪膏中黃芪甲苷含量的可靠性[J].中國臨床保健雜志,2014,17(6):577-579.
[11]劉楊,包華音,單成鋼.HPLC-ELSD法測定不同來源黃芪藥材中黃芪甲苷含量[J].山東中醫(yī)雜志,2014,33(12):1019-1021.
[12]曹桂萍,王曉晶.HPLC-ELSD法測定骨痹靈片中黃芪甲苷含量[J].現(xiàn)代中藥研究與實踐,2014,28(6):65-67.
[13]林楚迎.黃芪甲苷的提取工藝[J].科技創(chuàng)新導報,2014,11(35):229.
[14]李嘉華,陳奕伸,馮小映,等.雙香貼膏質量標準研究[J].中藥材,2014,37(11):2096-2098.
[15]王芳,尹文清,閔建國,等.壯藥紅根草的質量標準[J].中藥材,2014,37(11):1994-1997.
Content Determination of Astragaloside in Xiaokeping Tablets by RP-HPLCΔ
ZHOU Yingyi, LIU Xiaoxiao
(Dept.of Chinese Patent Medicine, Guangdong Institute for Drug Control, Guangdong Guangzhou 510180, China)
OBJECTIVE:To establish a method for the content determination of astragaloside in Xiaokeping tablets by RP-HPLC. METHODS: TLC method was used for the qualitative identification of coptidis rhizoma and puerariae lobatae radix. RP-HPLC was used to simultaneously determine the content of astragaloside in Xiaokeping tablets. The chromatographic column was Thermo BDS Hypersil C18(4.6 mm×250 mm,5 μm), the mobile phase was acetonitrile-water(V∶V=32 ∶68), with flow rate of 1 ml/min and column temperature of 30 ℃, and the detectors were evaporative light-scattering detector (ELSD). RESULTS: The qualitative identification was specific without interference in the negative reference. The linear range was 0.196 2-10.172 0 μg,r=0.999 8 for astragaloside. The average recovery rate was 100.7%,RSDwas 2.1% (n=9). CONCLUSIONS: The result of this study is accurate and reproducible, which can be used for quality control of Xiaokeping tablets.
Xiaokeping tablets; RP-HPLC; Astragaloside
2016-02-19)
廣東省省級科技計劃項目(No.201313040200029)
副主任藥師。研究方向:中藥質量標準提高。E-mail:cpulxx@126.com
R927.2
A
1672-2124(2016)07-0886-04
10.14009/j.issn.1672-2124.2016.07.008
*藥師。研究方向:藥品質量控制。E-mail:wingwa.2008@163.com
