夏枯草H PLC融合指紋圖譜研究
2016-08-04 07:27:59李敏陳蕾周倩王亮郭威
中國合理用藥探索 2016年4期
關(guān)鍵詞:融合
李敏 陳蕾 周倩 王亮 郭威
(1濟南市食品藥品檢驗檢測中心,山東 濟南250102;2深圳福田區(qū)中醫(yī)院,廣東 深圳518034;3山東中醫(yī)藥大學(xué)藥學(xué)院,濟南 250355;4山東省中醫(yī)藥研究院,濟南 250014)
夏枯草H PLC融合指紋圖譜研究
李敏1陳蕾2周倩3,4王亮4郭威3,4
(1濟南市食品藥品檢驗檢測中心,山東 濟南250102;2深圳福田區(qū)中醫(yī)院,廣東 深圳518034;3山東中醫(yī)藥大學(xué)藥學(xué)院,濟南 250355;4山東省中醫(yī)藥研究院,濟南 250014)
目的:建立夏枯草的融合指紋圖譜,實現(xiàn)同分異構(gòu)體熊果酸和齊墩果酸完全色譜分離和同步含量測定,對夏枯草質(zhì)量進(jìn)行評價。方法:采用高效液相色譜法,建立夏枯草的指紋圖譜,并為互為同分異構(gòu)體的熊果酸和齊墩果酸單獨建立分析方法,使其達(dá)到完全色譜分離,使用“MATLAB R2012a版”將兩個色譜圖融合部分進(jìn)行基線融合計算,使用“中藥色譜指紋圖譜相似度評價系統(tǒng)2004A版”進(jìn)行圖譜融合。結(jié)果:建立了可單獨、分段操作的“模塊式”融合指紋圖譜,共標(biāo)定了18個共有色譜峰,結(jié)合質(zhì)譜和對照品對部分共有峰進(jìn)行了推斷,其中1,3~ 8,17,18號峰分別推測為丹參素、咖啡酸、蘆丁、金絲桃苷、異槲皮苷、異迷迭香酸苷、迷迭香酸、齊墩果酸和熊果酸。同時對15批夏枯草樣品中熊果酸和齊墩果酸的含量進(jìn)行了測定,熊果酸含量0.19%~0.36%,齊墩果酸含量0.055%~ 0.11%。結(jié)論:建立的熊果酸和齊墩果酸的分析方法分離度高、分析速度快、結(jié)果準(zhǔn)確可靠;建立的“模塊式”融合指紋圖譜可以更準(zhǔn)確全面反映夏枯草中的化學(xué)成分,可為夏枯草質(zhì)量控制研究提供參考。
夏枯草;融合指紋圖譜;熊果酸;齊墩果酸;同分異構(gòu)體;高效液相色譜
夏枯草是臨床常見中藥,為唇形科植物夏枯草(Prunella vulgaris L.)的干燥果穗,有清肝瀉火,明目,散結(jié)消腫的功效[1]。現(xiàn)代藥理學(xué)研究表明,夏枯草的主要活性化學(xué)成分為三萜類、甾體類、黃酮類、香豆素類[2],其中熊果酸和齊墩果酸分別屬于烏蘇烷型和齊墩果烷型五環(huán)三萜,具有保肝、抗炎、抗病毒、抗微生物、抗糖尿病、抗腫瘤等作用,是夏枯草的主要有效成分[3-4],也是夏枯草質(zhì)量評價的重要含量測定指標(biāo),但是由于它們之間除1個甲基的取代位置略有不同外,化學(xué)結(jié)構(gòu)完全相同,因此很難分離[5-8]。指紋圖譜由于其可全面反映藥物中化學(xué)成分的種類和數(shù)量而作為重要的質(zhì)量評價方法得到廣泛的應(yīng)用,但其研究的側(cè)重點在于短時間內(nèi)反映大量成分的信息,因此即使指紋圖譜分析時間相對較長,結(jié)構(gòu)相近的同分異構(gòu)體同樣很難得到理想的分離效果。
目前融合指紋圖譜的研究多為不同檢測手段圖譜的融合[8],或因所含成分紫外吸收波長不一致而將不同波長的圖譜融合[9],基于同分異構(gòu)體的定量分析方法建立融合指紋圖譜的研究未見報道。為更加準(zhǔn)確、真實地鑒定夏枯草的質(zhì)量,本文擬采用高效液相色譜法(HPLC)建立夏枯草的指紋圖譜,并針對其中難以分離的熊果酸和齊墩果酸單獨建立特征分析方法,使用“MATLAB R2012a版”結(jié)合“中藥色譜指紋圖譜相似度評價系統(tǒng)2004A版”實現(xiàn)兩個圖譜的融合,以建立準(zhǔn)確反映夏枯草化學(xué)成分的融合指紋圖譜,同時對其中熊果酸和齊墩果酸兩種同分異構(gòu)體的含量進(jìn)行測定,以實現(xiàn)對夏枯草質(zhì)量更加客觀和準(zhǔn)確的評價。
1儀器與材料
1.1儀器
1200系列高效液相色譜系統(tǒng) (美國安捷倫公司),BP211D型電子天平(德國賽多利斯),Simplicity純水儀(美國密理博公司),LC-350A超聲波中藥處理機(濟寧市中區(qū)魯超儀器廠)。
1.2材料
熊果酸對照品(批號:X-006-140801-2),齊墩果酸對照品(批號:Q-003-140731-1),均購自成都瑞芬思科技生物有限公司,乙腈為色譜純,水為超純水,磷酸等其他試劑均為分析純,夏枯草為市售中藥飲片,經(jīng)山東省中醫(yī)藥研究院鑒定為唇形科植物夏枯草的干燥果穗。
2方法與結(jié)果
2.1對照品溶液的制備
取熊果酸和齊墩果酸對照品適量,精密稱定質(zhì)量,分別加甲醇制成每1 mL含熊果酸0.201 0 mg,含齊墩果酸0.180 0 mg的溶液,作為對照品儲備溶液。
2.2供試品溶液的制備
取各供試品粉末(過2號篩)0.25 g,精密稱定,精密加入甲醇25 mL,稱重,超聲提取30 min,冷卻至室溫,用甲醇補足減失的質(zhì)量,搖勻,濾過,取續(xù)濾液過0.45 μm微孔濾膜,即得。
2.3色譜條件
2.3.1夏枯草指紋圖譜色譜條件Agilent Eclipse XDB-C18(250 mm ×4.6 mm,5 μm)色譜柱;以乙腈(A)-0.1%磷酸水(B)為流動相;梯度洗脫(0~10 min,5% ~ 18%A;10~ 35 min,18%A;35~40 min,18%~30%A;40~55 min,30%A;55~ 75 min,30% ~ 100%A;75~ 100 min,100%A);檢測波長 210 nm;流速1.0 mL/min;柱溫30℃;進(jìn)樣量10 μL。色譜圖見圖1。
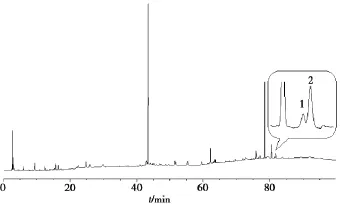
圖1 夏枯草指紋圖譜
2.3.2熊果酸和齊墩果酸色譜條件Agilent E-clipse XDB-C18(250 mm ×4.6 mm,5 μm)色譜柱;以乙腈-0.1%氨水(87∶13)為流動相;檢測波長210 nm;流速 1.0 mL/min;柱溫 30℃;進(jìn)樣量5 μL。色譜圖見圖2。
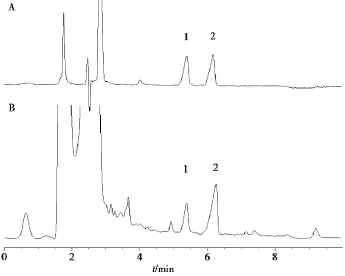
圖2夏枯草含量測定HPLC圖
2.4融合指紋圖譜的建立
使用“Chemstation B.01.03版”工作站將“2.3.1”和“2.3.2”中的色譜圖導(dǎo)出為AIA文件,使用“中藥色譜指紋圖譜相似度評價系統(tǒng) 2004A版”將AIA轉(zhuǎn)換為TXT格式,抽取兩個色譜圖中熊果酸和齊墩果酸的色譜峰數(shù)據(jù),使用“MATLAB R2012a版”將兩個色譜圖中熊果酸和齊墩果酸的數(shù)據(jù)進(jìn)行基線融合計算,將“2.3.1”中指紋圖譜中的對應(yīng)數(shù)據(jù)替換為融合計算后的數(shù)據(jù),得到的TXT文件導(dǎo)入“中藥色譜指紋圖譜相似度評價系統(tǒng)2004A版”得到融合指紋圖譜,見圖3。
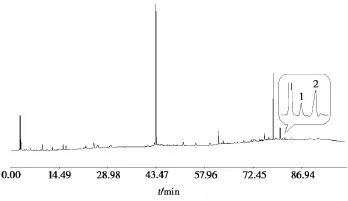
圖3 夏枯草融合指紋圖譜
2.5方法學(xué)考察
2.5.1精密度試驗取夏枯草樣品的供試品溶液,分別按照“2.3.1”和“2.3.2”中色譜條件連續(xù)測定6次,按照“2.4”中方法將色譜圖融合,以熊果酸的保留時間和峰面積為參照,分別對各共有峰相對保留時間和相對峰面積進(jìn)行統(tǒng)計。結(jié)果表明,各共有色譜峰的相對保留時間和相對峰面積基本一致,RSD均 <3%,表明儀器精密度良好。
2.5.2穩(wěn)定性試驗取夏枯草供試品溶液在室溫下保存,于 0,3,6,12,24 h分別按照“2.3.1”和“2.3.2”中色譜條件測定,按照“2.4”中方法將色譜圖融合,以熊果酸保留時間和峰面積為參照,分別對各共有峰相對保留時間和相對峰面積進(jìn)行統(tǒng)計。結(jié)果表明,各共有色譜峰的相對保留時間和相對峰面積基本一致,RSD均 <3%,表明樣品在24 h內(nèi)穩(wěn)定性良好。
2.5.3重復(fù)性試驗取夏枯草樣品(過2號篩)6份,按照“2.2”中方法制備供試品溶液,分別按照“2.3.1”和“2.3.2”中色譜條件依次測定,按照“2.4”中方法將色譜圖融合,以熊果酸保留時間和峰面積為參照,分別對各共有峰相對保留時間和相對峰面積進(jìn)行統(tǒng)計。結(jié)果表明,各共有色譜峰的相對保留時間和相對峰面積基本一致,RSD<3%,表明方法重復(fù)性良好。
2.5.4熊果酸和齊墩果酸線性和線性范圍考察精密吸取混合對照品儲備液,分別稀釋至儲備液濃度的0.025,0.05,0.1,0.3,0.5倍,制成熊果酸和齊墩果酸的系列標(biāo)準(zhǔn)溶液,按照“2.3.2”色譜條件進(jìn)行測定,測定峰面積,分別以峰面積積分值(y)對對照品濃度(x,mg/mL)進(jìn)行線性回歸處理,熊果酸的回歸方程為:
y=2 360.7x+4.95,
相關(guān)系數(shù)r為0.999 9,線性范圍為0.005 0~0.100 5 mg/mL;齊墩果酸的回歸方程為:
y=3 306.9x+2.158 3,
相關(guān)系數(shù)r為0.999 9,線性范圍為0.004 5~0.090 0 mg/mL。
2.5.5熊果酸和齊墩果酸加樣回收試驗取夏枯草樣品(過2號篩)6份,分別按樣品含量-對照品(1∶1)的大致比例加入一定量的對照品溶液,按照“2.2”中方法制備供試品溶液,按照“2.3.2”中方法進(jìn)行測定,計算回收率。結(jié)果測得熊果酸和齊墩果酸的回收率平均值分別為 97.16%和 98.72%,RSD分別為1.68%和 1.01%,表明方法測定結(jié)果準(zhǔn)確,見表1。
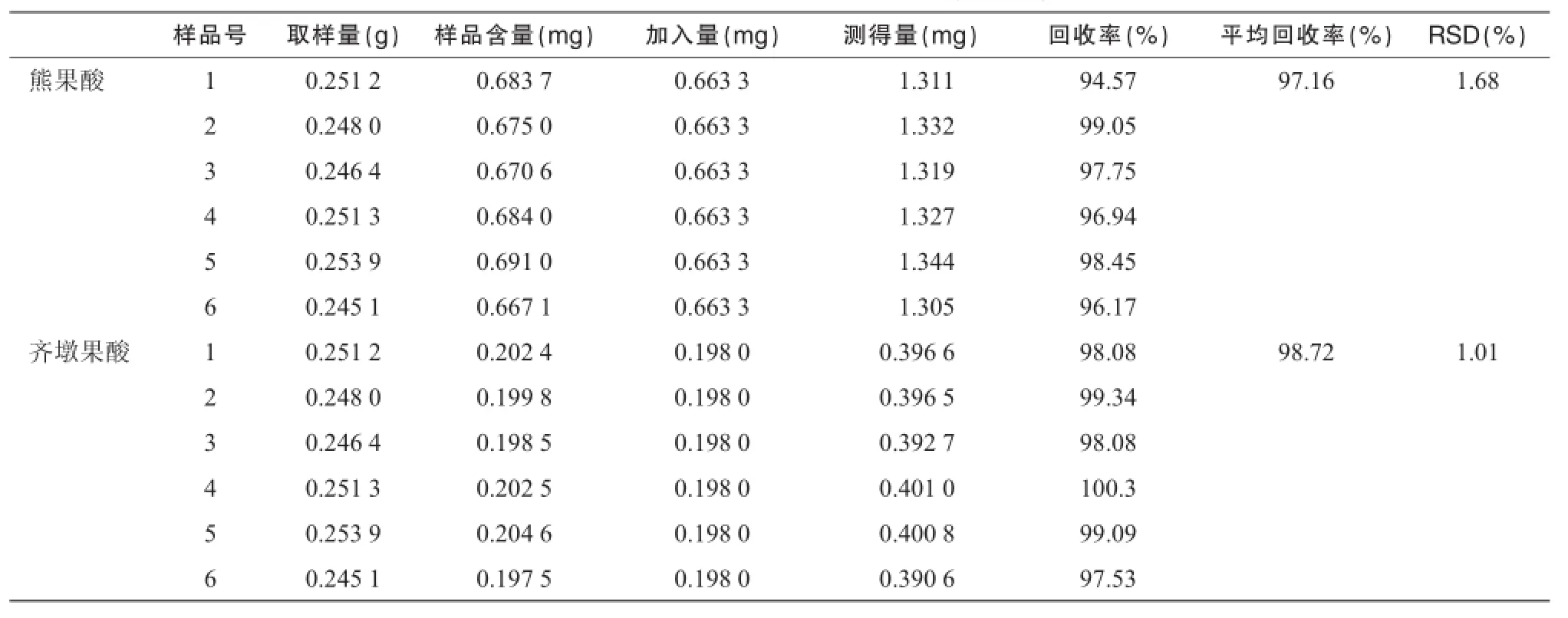
表1 夏枯草飲片回收率試驗結(jié)果(n=6)
2.6夏枯草飲片融合指紋圖譜共有模式的建立
將按照“2.4”中方法所得15批夏枯草融合指紋圖譜以TXT格式依次導(dǎo)入“中藥色譜指紋圖譜相似度評價系統(tǒng)2004A版”軟件。以1號夏枯草飲片色譜圖作為參照圖譜,經(jīng)過多點校正、自動匹配,以中位數(shù)法,生成對照圖譜,并標(biāo)定了18個共有色譜峰。夏枯草飲片HPLC融合指紋圖譜疊加圖和對照圖譜見圖4。
使用“中藥色譜指紋圖譜相似度評價系統(tǒng)2004A版”進(jìn)行相似度分析,15批次夏枯草與相應(yīng)對照圖譜的相似度均大于0.90,符合指紋圖譜要求。
2.7指紋圖譜質(zhì)譜分析
按照“2.3.1”中色譜條件,將磷酸替換為0.3%甲酸,對夏枯草樣品進(jìn)行電噴霧多級串聯(lián)質(zhì)譜(ESI-MSn)分析,對共有峰進(jìn)行了推測和推斷。由正負(fù)模式的準(zhǔn)分子離子峰可以推斷1,4,5,6,7,8號峰的分子量分別為 198,610,464,464,522和360 m/z,根據(jù)保留時間并結(jié)合文獻(xiàn)報道[10-12],推測1號峰為丹參素;結(jié)合對照品判斷3號峰為咖啡酸;4號峰裂解方式與文獻(xiàn)一致,推斷為蘆丁;5,6號峰裂解方式一致,一級質(zhì)譜均產(chǎn)生162 m/z中性丟失,剩余部分裂解方式與文獻(xiàn)報道中槲皮素一致,推斷為槲皮素的六碳糖苷類,結(jié)合夏枯草成分報道和保留時間數(shù)據(jù),推斷5,6號峰分別為金絲桃苷和異槲皮苷;推斷8號峰為迷迭香酸,7號峰發(fā)生162 m/z中性丟失后,剩余部分裂解方式與迷迭香酸相同,推斷7號峰為異迷迭香酸苷;結(jié)合對照品判斷 17,18號峰為齊墩果酸和熊果酸。見表2。
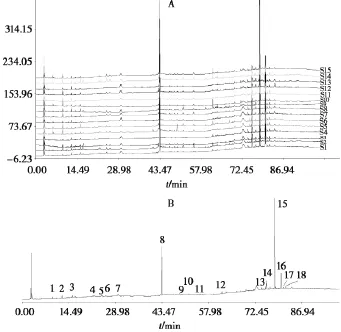
圖4 夏枯草飲片HPLC融合指紋圖譜疊加圖和對照圖譜
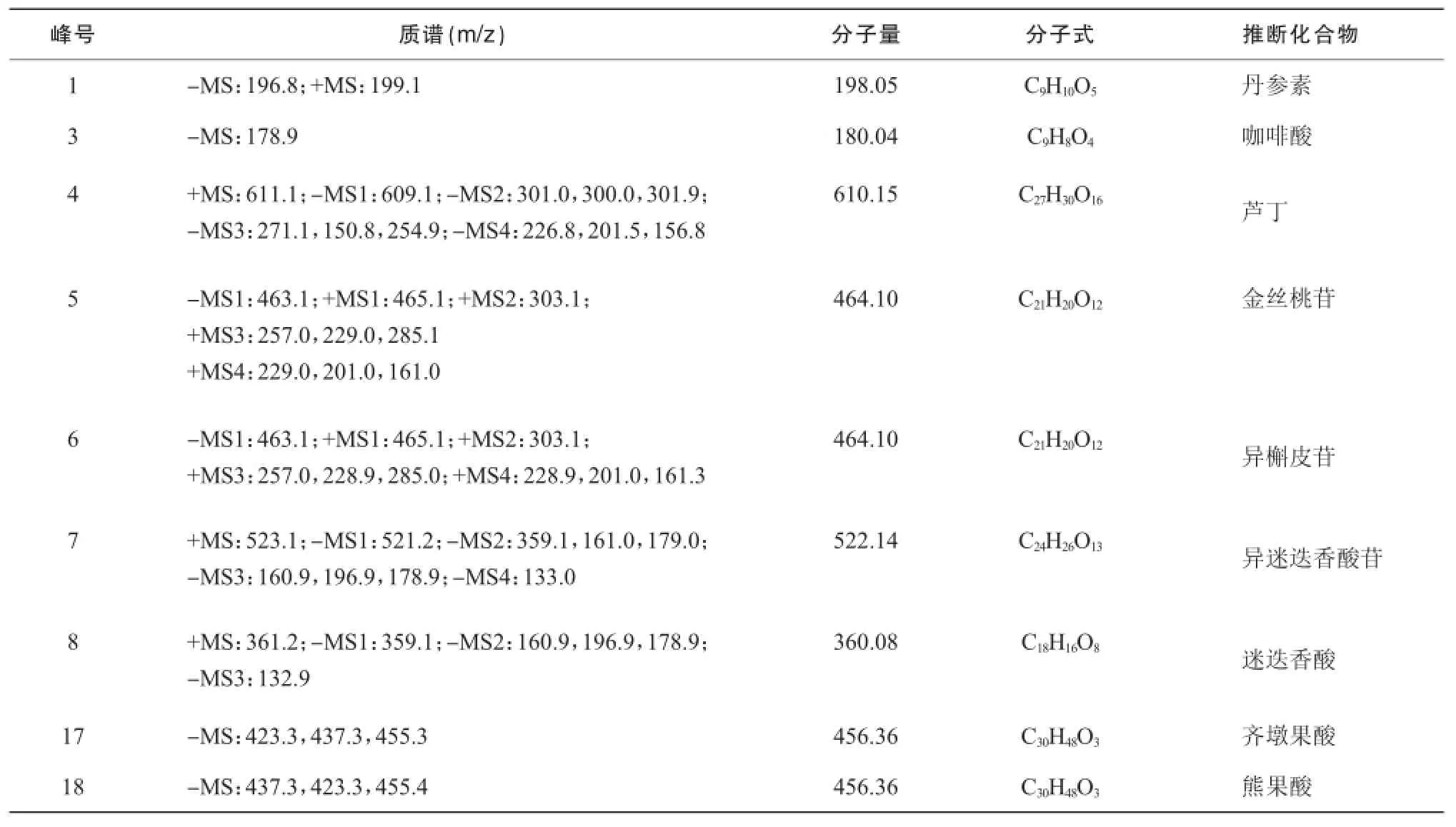
表2 夏枯草指紋圖譜質(zhì)譜分析
2.8主成分聚類分析
運用SPSS 19.0軟件中的因子分析對共有峰進(jìn)行標(biāo)準(zhǔn)化處理。以特征值>1為提取標(biāo)準(zhǔn),前5個成分的特征值大于1,分別為6.588,4.374,2.117,1.389,1.139,累計方差貢獻(xiàn)率達(dá)86.705%,能反映指紋圖譜主要信息,因此提取前5個成分進(jìn)行分析,計算主成分得分見表3。
由表 2中的主成分得分,運用SPSS 19.0軟件對15個批次夏枯草樣品采用組間平均數(shù)聯(lián)結(jié)法,以歐式平方距離對樣品聚類,得到聚類分析樹狀圖,見圖5。對樣品分類進(jìn)行判別分析得到區(qū)域圖,見圖6。
通過聚類分析可以將樣品分為3類,樣品1,3,5~ 13,15為一類;2,14為一類;4為一類。判別分析結(jié)果表明分類合理。從分析結(jié)果可以看出,夏枯草樣品總體質(zhì)量較為穩(wěn)定,少數(shù)批次成分差別較大,分類結(jié)果沒有明顯的地域特征。
2.9熊果酸和齊墩果酸含量測定
取夏枯草供試品溶液,按照“2.3.2”色譜條件進(jìn)行測定,測定峰面積,分別從標(biāo)準(zhǔn)曲線上計算出供試品溶液中熊果酸和齊墩果酸的濃度,計算樣品含量,結(jié)果見表3。

表3 夏枯草指紋圖譜主成分分析
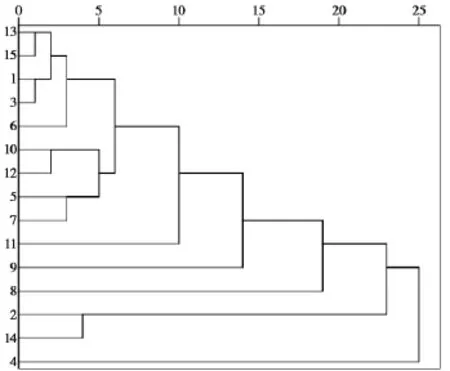
圖5 夏枯草系統(tǒng)聚類樹狀圖
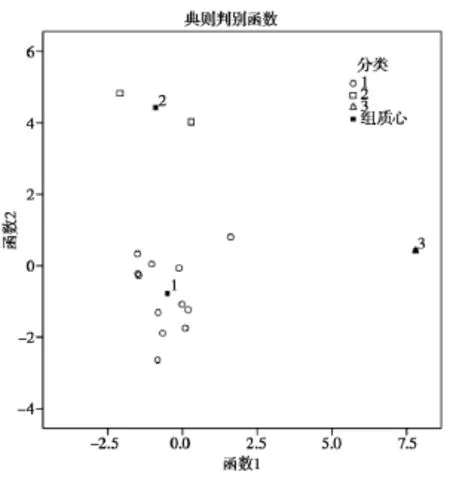
圖6 夏枯草判別分析區(qū)域圖
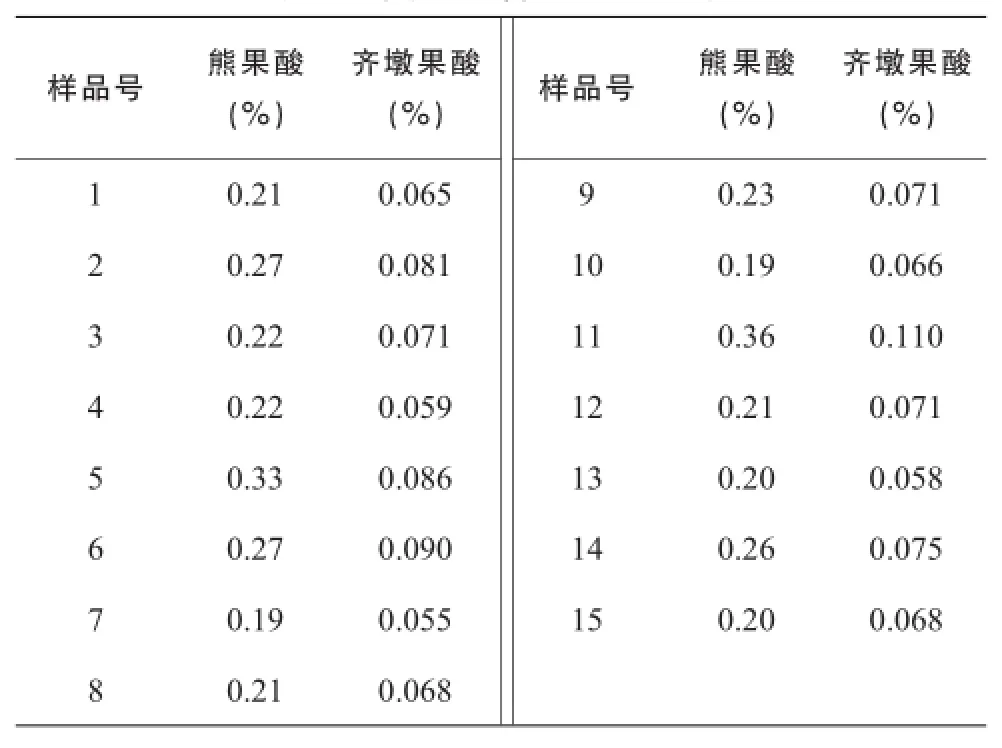
表4 夏枯草含量測定結(jié)果
3討論
夏枯草中熊果酸和齊墩果酸均為五環(huán)三萜類化合物,除1個甲基的取代位置略有不同外,化學(xué)結(jié)構(gòu)完全相同,因此分離一直是指紋圖譜研究的難點。二者含量測定雖已有文獻(xiàn)報道[5-8],但實驗室中重現(xiàn)不理想,很難達(dá)到基線分離,分離時間也較長,為15~ 45 min;且由于報道中均采用甲醇組成流動相,熊果酸和齊墩果酸的檢測波長為210 nm,接近甲醇的截止波長,導(dǎo)致基線不易平衡,信噪比很低。本研究中采用乙腈替代甲醇,很好地解決了信噪比低的問題;為改善二者分離度,同時在流動相中加入氨水,不足7 min即實現(xiàn)了兩種同分異構(gòu)體的基線分離,分離度達(dá)到 3.01。推測分離機理可能為,加入氨水后熊果酸和齊墩果酸中的羧基被離子化,成為強親水性基團,消除了羧基在色譜柱C18填料上的保留,從而提高了熊果酸和齊墩果酸中非極性部分在色譜柱C18填料上吸附的選擇性,進(jìn)而實現(xiàn)了色譜分離。再通過與傳統(tǒng)指紋圖譜的融合,很好地實現(xiàn)了快速、高效、全面評價飲片質(zhì)量的目的。
《中國藥典》2015年版用迷迭香酸含量評價夏枯草質(zhì)量,控制目標(biāo)單一,不能反映夏枯草的其他有效成分。本研究建立了夏枯草飲片的融合指紋圖譜,并同時對熊果酸和齊墩果酸進(jìn)行含量測定,各色譜峰分離度較好,基線較平穩(wěn),色譜信息較為豐富,主要色譜峰相對保留時間基本一致,而且色譜峰分布均勻,15個批次夏枯草共標(biāo)定了18個共有峰,可為夏枯草的質(zhì)量控制提供參考。通過對比,不同批次夏枯草指紋圖譜中共有峰相對保留時間較為一致,但相對峰面積有明顯差別,測得熊果酸的含量在0.19%~ 0.36%之間,齊墩果酸的含量在0.055% ~ 0.11%之間,成分含量差別較大,因此僅控制迷迭香酸含量并不能反映夏枯草的真實質(zhì)量,建議采用指紋圖譜對夏枯草進(jìn)行多成分同步質(zhì)量控制。
本研究將熊果酸和齊墩果酸的定量分析圖譜和夏枯草指紋圖譜進(jìn)行了融合,建立了“模塊式”的融合指紋圖譜,并結(jié)合質(zhì)譜技術(shù)部分消除了單一測定方法指紋圖譜的模糊性。在利用融合指紋圖譜對藥材進(jìn)行評價時,除了可以更全面、準(zhǔn)確地評價藥材中的化學(xué)成分外,對融合成分可以直接選用相應(yīng)的“模塊”色譜條件快速、準(zhǔn)確地進(jìn)行測定,不再需要采集整個指紋圖譜數(shù)據(jù),從而大大節(jié)省分析時間,可以預(yù)見,隨著研究的深入,融合指紋圖譜中的“模塊”越來越多,針對在實際應(yīng)用中不同的質(zhì)量控制要求會有越來越靈活高效的解決方案。融合指紋圖譜的整體性好,信息量豐富,分離度更優(yōu)化,可為真實、快速鑒別中藥材質(zhì)量提供參考,對于實際應(yīng)用具有重要意義。
[1] 國家藥典委員會.中華人民共和國藥典(一部)[S].北京:中國醫(yī)藥科技出版社,2015:280.
[2] 竇景云,于俊生.夏枯草藥理作用及臨床應(yīng)用研究進(jìn)展[J].現(xiàn)代醫(yī)藥衛(wèi)生,2013,29(7):1039-1041.
[3] 張明發(fā),沈雅琴.齊墩果酸和熊果酸的抗微生物和原蟲藥理研究進(jìn)展[J].抗感染藥學(xué),2010,7(3):153-156.
[4] 劉偉,丁海杰.HPLC測定夏枯草中熊果酸、齊墩果酸、迷迭香酸的含量[J].中成藥,2008,30(4):577-580.
[5] 邸學(xué),王海波,翟延君,等.HPLC測定藤梨根中熊果酸、齊墩果酸的含量[J].中國實驗方劑學(xué)雜志,2012,18(1):66-68.
[6] 張?zhí)m珍,巴寅穎,季思偉,等.RP-HPLC測定夏枯草不同部位熊果酸和齊墩果酸含量[J].藥物分析雜志,2009,29(9):1547-1549.
[7] 姚靜.HPLC測定不同產(chǎn)地澤蘭藥材中齊墩果酸和熊果酸含量[J].中國實驗方劑學(xué)雜志,2015,21(10):80-82.
[8] 李全斌,何開勇.齊墩果酸含量測定的研究進(jìn)展[J].中國執(zhí)業(yè)藥師,2011,8(7):32-34.
[9] 崔媛,王小明,楊勇,等.薏苡仁油融合指紋圖譜研究[J].中草藥,2014,45(12):1698-1701.
[10] 方芳,王晶,方舟,等.復(fù)方黃白膠囊的多波長融合HPLC指紋圖譜[J].中國實驗方劑學(xué)雜志,2013,19(17):113-117.
[11] 梁杰康,張琳,嚴(yán)曉明.HPLC-ESI-MS/MS鑒定夏枯草的主要化學(xué)成分[J].中國中醫(yī)藥現(xiàn)代遠(yuǎn)程教育,2013,11(14):153-154.
[12] 封亮,賈曉斌,陳彥,等.夏枯草化學(xué)成分及抗腫瘤機制研究進(jìn)展[J].中華中醫(yī)藥雜志,2008,23(5):428-434.
[13] 袁麗春,劉斌,石任兵.HPLC法測定不同市售荷葉藥材中金絲桃苷和異槲皮苷的含量[J].藥物分析雜志,2010,30(1):41-44.
Study on Fusion Fingerprint of Prunellae Spica by HPLC
Li Min1,Chen Lei2,Zhou Qian3,4,Wang Liang4,Guo Wei3,4
(1 Jinan Center for Food and Drug Control,Shandong Jinan 250102,China;2 Shenzhen Futian Hospital of TCM,Guangdong Shenzhen 518034;3 School of Pharmaceutical Sciences,Shandong University of Traditional Chinese Medicine,Jinan 250355;4 Shandong Academy of Traditional Chinese Medicine,Jinan 250014)
Objective:To establish the fusion fingerprint of prunellae spica,perform the complete chromatographic separation and synchronous content determination of two isomers,ursolic acid and oleanolic acid,and evaluate the quality of prunella spica.Methods:The fingerprint of prunellae spica was established by HPLC,and a special assay for ursolic acid and oleanolic was established for complete chromatographic separation.The baselines of the fusion part in the two chromatograms were calculated and corrected by“MATLAB R2012a”,and the chromatograms were fused by“Similarity Evaluation System for Chromatographic Fingerprint of TCM 2004A”.Results:A“modular”fusion fingerprint which could be performed individually and partly was established,in which18 common peaks were included.Thepeaks 1,3~ 8,17 and 18 were characterized by MS and standard samples,which were danshensu,caffeic acid,rutinum,hyperoside,isoquercitrin,salviaflaside,rosmarinic acid,oleanolic acid and ursolic acid,respectively.The determination results of ursolic acid and oleanolic acid contents in 15 batches of prunellae spica were 0.19%~0.36%and 0.055%~0.11%respectively.Conclusion:The developed method for determination of ursolic acid and oleanolic acid showed a high separation resolution,which was rapid,accurate and reliable.The established“modular”fusion fingerprint reflected the chemical composition of prunellae spica comprehensively and accurately,which provided a reference for the study on quality control of prunella spica.
Prunellae Spica;Fusion Fingerprint;Ursolic Acid;Oleanolic Acid;Isomers;HPLC
10.3969/j.issn.1672-5433.2016.04.006
深圳市福田區(qū)衛(wèi)生公益性科研項目(FTWS2014028);山東省中醫(yī)藥科技發(fā)展計劃(2015-167)
李敏,女,工程師。研究方向:中藥檢驗和中藥質(zhì)量控制。E-mail:568027183@qq.com周倩,女,碩士,助理研究員。主要從事中藥炮制研究。通訊作者E-mail:merveilleqq@163.com
2012-12-25)
猜你喜歡
中學(xué)生數(shù)理化·中考版(2022年8期)2022-06-14 06:55:24
數(shù)學(xué)年刊A輯(中文版)(2022年4期)2022-02-16 08:17:34
今日農(nóng)業(yè)(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
無線電通信技術(shù)(2021年4期)2021-07-13 08:58:28
無線電通信技術(shù)(2021年3期)2021-06-08 03:33:48
中學(xué)生數(shù)理化(高中版.高考數(shù)學(xué))(2021年1期)2021-03-19 08:28:38
無線電工程(2020年11期)2020-10-29 01:25:46
現(xiàn)代出版(2020年3期)2020-06-20 07:10:34
福利中國(2015年4期)2015-01-03 08:03:38
