環戊烷-甲烷水合物生成過程的溫度特性
2016-07-14 09:46:09胡亞飛蔡晶李小森中國科學院廣州能源研究所中國科學院天然氣水合物重點實驗室廣東省新能源和可再生能源研究開發與應用重點實驗室廣東廣州50640中國科學院大學北京00049
化工進展 2016年5期
胡亞飛,蔡晶,李小森(中國科學院廣州能源研究所,中國科學院天然氣水合物重點實驗室,廣東省新能源和可再生能源研究開發與應用重點實驗室,廣東 廣州 50640;中國科學院大學,北京 00049)
?
研究開發
環戊烷-甲烷水合物生成過程的溫度特性
胡亞飛1,2,蔡晶1,李小森1
(1中國科學院廣州能源研究所,中國科學院天然氣水合物重點實驗室,廣東省新能源和可再生能源研究開發與應用重點實驗室,廣東 廣州 510640;2中國科學院大學,北京 100049)
摘要:研究了環戊烷-甲烷水合物生成過程中的溫度變化,分析了體系的熱量損失。在不同初始溫度(4℃、8℃和12℃)、壓力(2MPa、4MPa、6MPa、8MPa和10MPa)和進氣方式(一次性進氣、連續進氣和間歇進氣)的條件下,測定了釜內溫度,對比了以上各因素對釜內最高溫度(Tmax)與釜內最大溫升(ΔTmax)的影響。實驗表明,Tmax主要受壓力和進氣方式影響,初始溫度對其影響不明顯;ΔTmax受初始溫度、壓力和進氣方式影響顯著。在間歇進氣方式下,初始溫度越低、壓力越高,ΔTmax越大。其中,在初始溫度為4℃、壓力為10MPa、進氣時間間隔為30min的間歇進氣方式下,ΔTmax可達16.5℃。此外,由熱量分析發現,體系的主要熱量損耗表現為體系向環境中的散熱。因此,提高保溫層的絕熱性能,有利于提高水合物生成熱的熱量有效利用率。
關鍵詞:水合物;甲烷;環戊烷;生成熱;最大溫升
第一作者:胡亞飛(1989—),男,碩士研究生。聯系人:李小森,博士,研究員,博士生導師,主要從事天然氣水合物方面的研究。E-mail lixs@ms.giec.ac.cn。
氣體水合物是由輕烴、二氧化碳、硫化氫等氣體小分子與水分子在低溫或高壓條件下形成的一種非化學計量的籠型包絡化合物[1]。其中,主體水分子通過氫鍵構建出籠型孔穴結構,客體分子填充于孔穴結構之中,通過范德華力與水分子結合,從而形成結構穩定的水合物。氣體水合物的形成是一種特殊的相變過程,在水合物形成過程中物質由氣相或液相轉移到水合物固相,并伴隨著相變熱的釋放。根據水合物的相變特性,衍生出了一系列的水合物應用技術,如利用水合物進行海水淡化[2-3]、二氧化碳(CO2)捕獲與封存[4-5]以及將水合物作為蓄冷工質應用于空調蓄冷系統中等[6-7]。其中,水合物在空調蓄冷技術中的應用是基于水合物形成和分解過程中的相變熱特性。然而,由于純氣體水合物形成條件苛刻、水合物生成時間普遍較長,導致氣體水合物形成過程中釋放的水合物相變熱熱量有限且熱量損耗嚴重。為提高水合物相變熱熱量的釋放,通常采用添加水合物促進劑加快水合物形成速率的方式來增加水合物相變熱的釋放,降低單位時間內的熱量損失。常見的熱力學促進劑通過參與水合物結構的構建、降低形成水合物的相平衡條件來加速水合物的形成;動力學促進劑通過降低溶液的表面張力、增加氣液接觸面積的方式來加速水合物的形成速率,從而增加水合物相變熱的釋放量。相關合適的水合物熱力學促進劑主要有環戊烷(CP)、四丁基溴化銨(TBAB)和四氫呋喃(THF)等[8-12],動力學促進劑有十二烷基硫酸鈉(SDS)[13]和十二烷基苯磺酸鈉(SDBS)[14]等。
根據相變熱在氣體水合物形成與分解過程中的釋放特點,將水合物形成過程中釋放的熱量稱為水合物生成熱,將水合物分解過程中吸收的熱量稱為水合物分解熱。對水合物形成分解過程中相變熱的定量分析一直是水合物熱特性研究的熱點和難點。目前,水合物相變熱數值的確定主要有兩種方法,分別為:直接法——利用差示掃描量熱儀(DSC)實驗測定水合物分解熱[15-17];間接法——利用Clausius-Clapeyron方程計算獲得水合物分解熱[18-19]。其中,直接法為實驗方法,借助DSC實驗設備測定相應條件下水合物形成或分解過程中的熱流變化,從而獲得對應水合物的理論相變熱數值;而間接法為理論計算方法,在水合物相平衡數據的基礎上,根據Clausius-Clapeyron方程計算得到理論相變熱數值?H。然而,以上兩種確定水合物相變熱的方法都只從水合物熱特性參數的基礎研究出發,而非出于水合物相變熱熱量實際應用價值的研究目的,尤其針對保溫系統中,因水合物生成熱釋放而引起的體系溫度特性研究較少。中國科學院天然氣水合物重點實驗室李小森課題組[20-21]首次提出了一種直接利用水合物生成熱加熱海水形成熱鹽水進行天然氣水合物原位開采的方法,結果表明該方法相對傳統的注熱鹽水開采法具有明顯的優越性,拓展了氣體水合物相變熱的應用領域。其中水合物形成過程中生成熱所引起的體系溫度變化是影響該水合物原位開采方法開采效率的重要因素之一。
因此,本文以釜內溫度變化為考察對象,針對環戊烷-甲烷水合物的形成體系,分別考察了不同初始溫度、壓力以及進氣方式對環戊烷-甲烷水合物生成過程中釜內溫度變化的影響,進而分析其對釜內最高溫度以及釜內最大溫升的影響,并對水合物生成體系的熱量損失進行了詳細的分析。
1 實驗部分
1.1 實驗材料
實驗所用氣體為摩爾分數 99.9%的純甲烷氣體,由佛山華特氣體有限公司提供;環戊烷純度為99.0%,由成都貝斯特試劑有限公司提供;實驗用水為電導率18.25m?/cm的去離子水,由南京超純水技術有限公司提供的超純水設備制備。
1.2 實驗裝置
圖1為水合物生成過程中溫度測定的實驗裝置示意圖,該裝置主要由進排液模塊、供氣模塊、反應釜、低溫室以及數據采集模塊五部分組成。反應釜由 316不銹鋼制成,釜高為 100mm,釜內徑為40mm,釜內總體積為125mL,最大承壓為15MPa。如圖1所示,釜內自上往下分布有3個Pt100熱電偶,其測量值分別為T1、T2、T3,溫度的測量精度為±0.01℃。反應釜頂部安置有型號為trafag8251的壓力傳感器,用以測量反應釜內壓力,測壓范圍為0~40MPa,測量精度為±0.02MPa。反應釜釜體用熱導率較小的氣凝膠保溫材料[λ<0.02W/(m·K),25℃時]進行包裹,氣凝膠層的包裹厚度為50mm。供氣模塊中高壓貯氣罐總體積為 8L,最大承壓為45MPa,頂部安置有Pt100熱電偶和型號為setra5310的壓力傳感器,測壓范圍為 0~40MPa,測量精度為±0.02MPa。低溫室溫度由氟利昂吊頂式冷風機進行控制調節,其溫度通過Pt100熱電偶監控,記錄為Te。其中,實驗中的所有壓力和溫度值由計算機軟件自動采集并記錄。

圖1 實驗裝置示意圖
1.3 實驗方法
環戊烷-甲烷水合物是結構 II型水合物[22],理想比例構成的環戊烷-甲烷水合物的分子表達式為8CP·16CH4·136H2O。因此,實驗采用環戊烷與水的摩爾比例為1∶17,按反應液體積為70mL轉換為體積,則 CP與 H2O的體積分別為 16.5mL和53.5mL,本文所有實驗均在此比例下進行,去離子水與環戊烷的混合反應液體積均為70mL。
水合物生成實驗開始前,調節低溫室溫度為實驗設定值,實驗中需要使用的氣體與液體均放置于低溫室中,保證實驗前得到充分冷卻。實驗開始前,用增壓泵將貯氣罐中甲烷氣增壓至30MPa。使用真空泵對反應釜抽真空,采用負壓注入去離子水清洗反應釜3次。隨后對反應釜再次抽真空,將環戊烷與水的混合反應液注入反應釜,靜置 10min。之后從釜底鼓泡通入甲烷氣體至實驗設定壓力,測定并記錄釜內溫度變化。實驗中分別考察了初始溫度為4℃、8℃和12℃,壓力為2MPa、4MPa、6MPa、8 MPa和10MPa,以及進氣方式分別為一次性進氣、連續進氣和間歇進氣對釜內溫度的影響。實驗過程中,儲氣罐中出口處甲烷氣的壓力通過調節調壓閥維持恒定。當儲氣罐內的壓力保持30min不變化時,則認為該條件下的水合物生成過程結束。水合物生成實驗結束后,打開釜頂控制閥排空釜內自由氣,并將反應釜放置于20℃的條件下分解水合物,利用排水集氣法確定水合物分解相中甲烷氣的體積。最后,排出釜內反應液,清洗反應釜并將反應釜移至低溫室,調節低溫室溫度為實驗設定溫度,重復進行下一組實驗。
2 結果與討論
2.1 初始溫度的影響
在壓力為10MPa、進氣方式為連續進氣的實驗條件下,討論了不同初始溫度T0(4℃、8℃和12℃)對釜內溫度T1、T2、T3的影響,確定不同初始溫度條件下的釜內最高溫度和釜內最大溫升。在水合物生成過程中將測定的反應釜內最高溫度稱為釜內最高溫度(Tmax),釜內最高溫度與初始溫度之差為釜內最大溫升(ΔTmax)。Te為低溫室溫度,在±1.5℃的溫度范圍內波動。
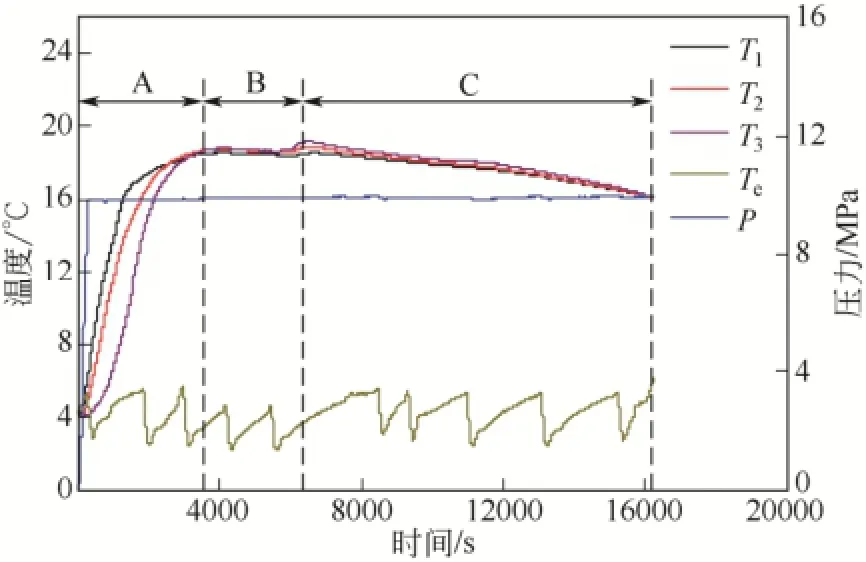
圖2 初始溫度為4℃、壓力為10 MPa條件下釜內溫度T1、T2、T3的變化圖
圖2為初始溫度為4℃、壓力為10 MPa時,水合物生成過程中釜內溫度和壓力隨水合物生成時間的變化圖。如圖2所示,在水合物生成過程中,釜內溫度出現了 3種不同的變化趨勢:在 0~60min內,釜內溫度快速上升,如A段所示;在60~105min內,釜內溫度維持穩定,如B段所示;105~270min內,釜內溫度不斷下降,如C段所示。如圖2所示,A段中釜內不同位置的溫度T1、T2、T3均由4℃快速上升至18.8℃附近。由此可見,隨著甲烷氣體從反應釜底部不斷地鼓入,釜壓從0增至實驗壓力10MPa,環戊烷-甲烷水合物大量生成,在生成過程中伴隨著大量生成熱的釋放。同時,由于本實驗使用的反應釜(如圖1所示)包裹有保溫層,使得水合物生成過程中釋放的相變熱累積在反應體系中,致使釜內溫度不斷升高,從而實現加熱整個反應體系的目的。在 A段水合物生成過程中,T1、T2、T3瞬時速率均呈現出先增加后降低的變化趨勢。這是由于大量生成的水合物不斷地在氣液界面堆積,減小了水合物形成過程中的氣液接觸面積,與此同時,受釜內溫度上升的影響,新水合物形成驅動力降低,水合物生成速率隨之減慢。然而,不斷有氣體鼓入釜內,氣體從反應釜底部上升到氣液界面,此過程中的氣體擾動作用對氣液接觸面的更新具有一定的積極作用。在以上3個抗衡因素的作用下,T1、T2、T3瞬時速率會不斷發生變化。A段前期,水合物在氣液界面還未形成明顯的堆積,此時氣液接觸面積大且氣體擾動致使氣液接觸面更新較快,因而釜內溫度瞬時速率大;隨著水合物在氣液界面上不斷地堆積,水合物的生成因氣液接觸面的更新受限和釜內溫度升高的雙重影響,從而使得釜內溫度瞬時速率不斷降低,直至B段,釜內溫度瞬時速率為零。
此外,對比圖2中A段T1、T2、T3各自的溫度變化發現,相同時間點釜內溫度呈現出T1> T2> T3的變化特點。由圖1可知,T1溫度傳感器位于氣液界面附近,T2、T3溫度傳感器均位于溶液中。綜合圖1與圖2可知,環戊烷-甲烷水合物最初在氣液界面處生成,隨著反應的不斷進行,水合物生成界面不斷地向溶液中移動。因此,水合物生成過程中釋放的大量相變熱首先加熱 T1,隨后依次加熱 T2和T3。在釜壓維持恒定壓力15min后,T1為15.5℃,T3為 8.0℃,此時 T1與 T3間的溫差值最大,高達7.5℃。A段后期,釜內溫度T1、T2、T3逐漸接近,最后均達到了18.8℃。此時,體系中氣液接觸面更新受限、水合物生成驅動力減小,環戊烷-甲烷水合物生成速率減慢。當單位時間內水合物生成熱釋放熱量與反應釜損失熱量相近時,釜內溫度T1、T2、T3開始穩定,從而轉為B段的釜內溫度穩定階段。B段中,由于底部鼓入的氣體與水合物堆積層中間隙水之間存在著一定的有效氣液接觸面,因此,此階段中仍有少量水合物生成。然而,受釜內溫度高和反應釜熱量損失的影響,釜內溫度T1、T2、T3變化不明顯。隨著反應釜內水合物在堆積層中不斷生成,水合物漿液越來越致密,氣液接觸越來越困難,使得水合物生成過程中的傳質阻力不斷增大,從而導致水合物生成速率減慢甚至停止。水合物生成熱釋放熱量小于反應釜向低溫環境散失的熱量,釜內溫度T1、T2、T3由穩定的B階段進入下降的C段。圖3為壓力為10MPa,初始溫度分別為4℃、8℃、12℃時,以釜內溫度T2為例詳述水合物生成過程中釜內溫度隨時間的變化圖。從圖3可知,在初始溫度為8℃和12℃條件下,釜內溫度T2的溫度變化趨勢與圖2所示的釜內溫度T1、T2、T3的變化趨勢相同,都經歷了圖2所示的A、B、C三段不同的溫度變化過程。然而,在初始溫度為12℃的條件下,釜內溫度上升段(A段)持續的時間長達165 min,遠大于初始溫度為 4℃和 8℃條件下 60min和64min。并且在初始溫度為12℃條件下的A段前期,釜內溫度變化較小且呈現出明顯的溫度平臺,而在A段后期,釜內溫度變化明顯且溫升速率快。由圖3可知,在初始溫度為12℃條件下氣體初始通入反應釜便出現第一段溫升(0~15min),該溫升較小,釜內溫度達 13.4℃后便由溫升轉為溫降,經過了100min的緩慢溫降后,釜內溫度降至13.1℃;隨后出現第二段溫升,第二段溫升(105~165min)結束時釜內溫度升至20.6℃。引起第一段溫升的熱量主要來源為甲烷氣在反應液中的溶解熱,而引起第二段溫升的熱量來源為大量環戊烷-甲烷水合物生成所釋放的相變熱。在初始溫度為12℃時,水合物的形成驅動力下降,形成水合物需要較長的誘導時間,因此在A段前期出現了一段較長時間的溫度平臺,且釜內溫升僅為1.1℃。在4℃和8℃初始條件下,因初始溫度降低,體系中形成水合物的溫度驅動力增大,水合物形成的誘導時間縮短,A段釜內溫度 T2呈現快速上升的變化趨勢。此外,從圖 3可知,在初始溫度為12℃條件下Tmax達20.6℃,高于初始溫度為4℃和8℃條件下的Tmax達18.8℃和20.0℃。這可能是因為初始溫度提高后,對應的低溫環境溫度較高,在生成水合物的過程中體系向低溫環境中散失的熱量減少所致。
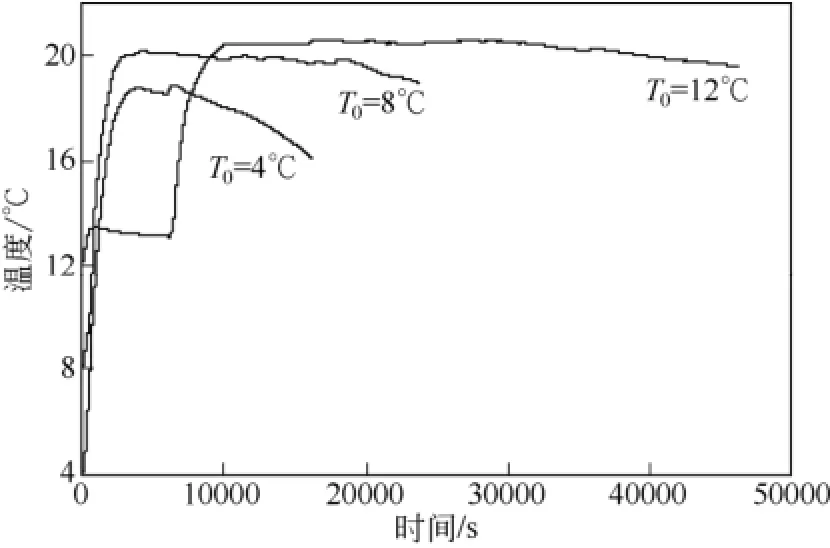
圖3 壓力為10 MPa,初始溫度分別為4℃、8℃、12℃條件下釜內溫度T2的對比圖
根據相平衡曲線可知,在10MPa壓力條件下的環戊烷-甲烷水合物體系中,理想的釜內最高溫度Tmax值為該相平衡點的溫度,約為30℃[23]。然而,在實際的實驗過程中,受釜內溫度升高、水合物生成速率減慢和體系熱量損失的影響,致使反應體系中Tmax存在溫度上限,該上限值小于30℃,同時反應體系中存在ΔTmax。圖4為實驗壓力為10MPa的恒壓條件下,初始溫度不同所引起的ΔTmax以及甲烷氣體消耗量的對比圖。從圖4可知,相同壓力條件下初始溫度越高,ΔTmax越小,ΔTmax值由初始溫度為4℃條件下的14.8℃降低到初始溫度為12℃條件下的 8.6℃。即降低反應初始溫度,有利于提高ΔTmax。如圖4所示,初始溫度為4℃、8℃和12℃條件下,甲烷氣體的消耗量分別為5.66L、5.24L和4.60L,即初始溫度越高,甲烷氣體消耗量越小。甲烷氣體的消耗量越小,說明環戊烷-甲烷水合物的生成量越小,水合物生成過程中釋放的相變熱熱量越小,影響著ΔTmax數值的大小。
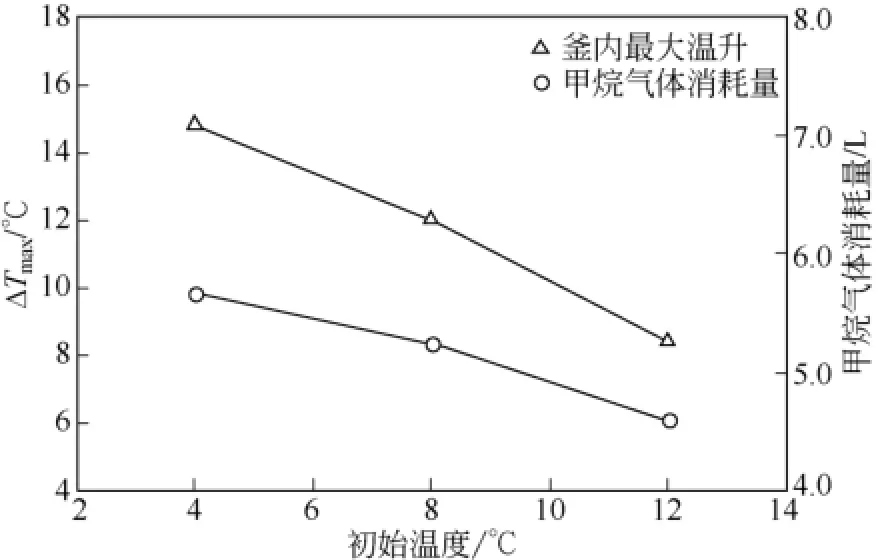
圖4 初始溫度分別為4℃、8℃、12℃條件下的釜內最大溫升ΔTmax和甲烷氣體消耗量對比圖
2.2 壓力的影響
在初始溫度為 4℃、進氣方式為連續進氣的條件下,討論了不同壓力(2MPa、4MPa、6MPa、8MPa 和10MPa)對釜內溫度T1、T2、T3的影響,對比不同壓力條件下的Tmax和ΔTmax值。圖5為反應壓力分別為2MPa、4MPa、6MPa、8MPa和10MPa以及連續進氣條件下的釜內溫度變化圖。從圖5可知,不同壓力條件下,各體系的溫升時間起點以及釜內溫度達到 Tmax所需時間不同,呈現出低壓(2MPa 和4MPa)和高壓(6MPa、8MPa和10MPa)兩種不同的變化特點。在2MPa和4MPa的低壓條件下,從反應開始到釜內溫度出現明顯變化至少需要17h,水合物形成誘導時間長,且Tmax分別僅為5.6℃和12.6℃;而在6MPa、8MPa和10MPa的較高壓力條件下,水合物形成誘導短,均在4h之內就完成了如圖2所示的A段釜內溫度快速上升階段,且Tmax分別達到了 16.8℃、17.8℃和 18.8℃。由此可見,壓力對釜內溫度的影響顯著。相同初始溫度條件下,壓力越高,水合物的形成驅動力越大,水合物生成速率越快,水合物的生成量越大,越能有效地提高體系的Tmax值。由圖5可知,低壓條件下反應釜內溫度的升高呈現了3個不同的變化階段,分別表現為:①0~15min,溫度變化速率快但變化幅度較小,熱量來源于大量甲烷氣溶解于反應液中釋放的溶解熱;②1~17h,溫度變化速率緩慢且變化幅度不大,熱量來源于形成少量水合物時所釋放的水合物生成熱;③17~25h,溫度變化速率快且變化幅度較大,熱量來源于釜內快速形成大量水合物時釋放的大量生成熱。從圖5中較高壓力條件下釜內溫度變化曲線可知,反應釜內溫度的升高僅呈現了類似于圖2中A段的溫升變化。在6MPa、8MPa 和 10MPa壓力條件下對慶的溫升時間分別分為235min、96min和60min,此時Tmax維持穩定的時間(即對應圖2中B段的時間)分別為241min、47min 和45min。由圖5可知,6MPa壓力條件下Tmax維持穩定的時間最長。這是因為6MPa壓力下生成的水合物致密程度較低,水合物堆積層中有一定量間隙水存在,在壓力穩定的情況下,間隙水逐漸轉化為水合物,持續釋放出少量相變熱,同時,與8MPa和10MPa壓力條件相比,6MPa壓力下的Tmax與低溫環境間的溫差較小,釜內熱量向低溫環境中散失的速率低。
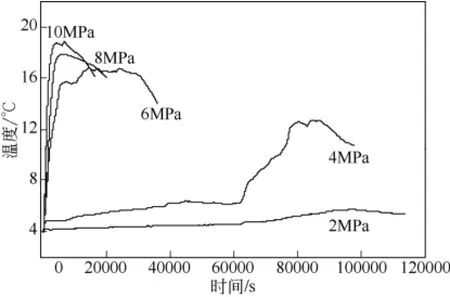
圖5 初始溫度為4℃、不同壓力條件下釜內溫度T2的變化對比圖
從圖6可知,隨著壓力的升高,ΔTmax呈現遞增的趨勢,但增加的幅度隨著壓力的升高而降低。在4MPa、8MPa與10MPa 3種壓力條件下,甲烷氣體消耗量相近,但ΔTmax差異明顯。這是因為甲烷氣體消耗量相近時,壓力越高,生成水合物的壓力驅動力越大,水合物生成速率越快,Tmax越高,ΔTmax也越大。在6MPa壓力條件下,環戊烷-甲烷水合物的甲烷氣體消耗量為7.04L,大于8MPa和10MPa壓力條件下甲烷氣體消耗量5.36L和5.66L。這是因為在6MPa壓力下,當釜內溫度達Tmax時,因水合物致密程度低,此時有大量間隙水轉化到環戊烷-甲烷水合物中,生成了更多的環戊烷-甲烷水合物。由圖6可知,6MPa、8MPa與10MPa壓力條件下的ΔTmax分別為12.8℃、13.8℃和14.8℃,即壓力越高,ΔTmax越大。由此可見,壓力、水合物生成量以及散熱速率都影響著釜內溫升的大小,其中壓力對環戊烷-甲烷水合物生成過程中釜內溫升的影響最為顯著。
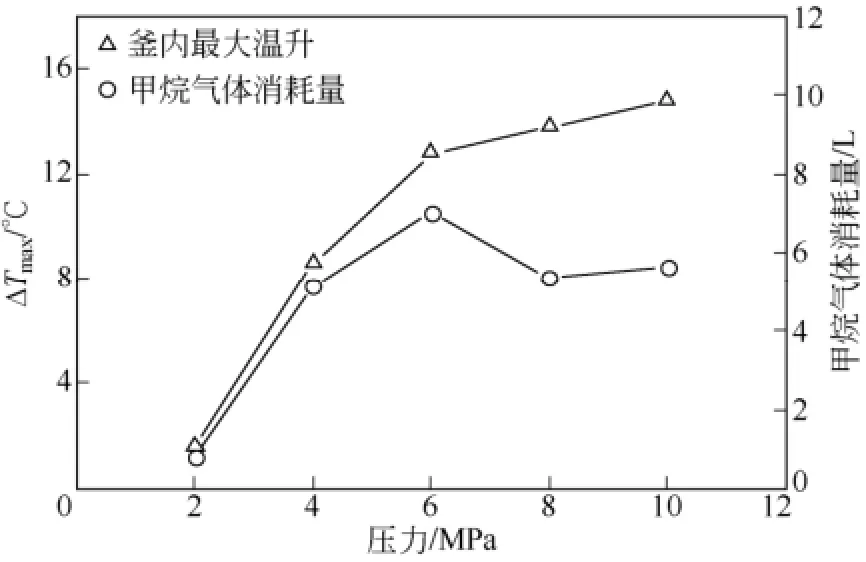
圖6 初始溫度為4℃、不同壓力條件下的釜內最大溫升ΔTmax和甲烷氣體消耗量對比圖
2.3 進氣方式的影響
在初始溫度為4℃、壓力為10MPa,討論了不同進氣方式對釜內溫度T1、T2、T3的影響,確定不同進氣方式條件下的Tmax和ΔTmax。對于不同進氣方式實驗,進氣方式有一次性進氣、連續進氣和間歇進氣3種。其中,一次性進氣方式為直接增壓至實驗壓力后停止進氣;連續進氣方式為直接增壓至實驗壓力后維持實驗壓力恒定的持續進氣;間歇進氣方式為分段增壓進氣至實驗壓力,其中,分段壓力采用2MPa、4MPa、6MPa、8MPa、10 MPa,本實驗進氣間隔采用10min和30min兩種。
2.3.1 一次性進氣方式與連續進氣方式
圖7為一次性進氣下體系的釜內溫度和壓力變化圖。根據釜內壓力的變化過程,可將釜內壓力劃分為釜壓急劇上升段(a段)、釜壓下降段(b段)、釜壓快速下降段(c段)和釜壓緩慢下降段(d段)。由圖7可知,釜內溫度在b段幾乎不變化,在c段時,因水合物大量生成,釜內熱量集中,釜內溫度快速增加至12.1℃。釜內溫度達12.1℃后,釜內壓力下降明顯變慢(d段),但釜內溫度仍在緩慢升高,此時釜壓下降的原因主要為釜內水合物仍在緩慢生成而不斷消耗甲烷氣體;釜溫升高至最高溫度12.5℃后,釜內溫度由溫升轉為溫降,此時釜壓緩慢下降的主要為:因釜內溫度下降,由氣體狀態方程可知釜壓也會同步下降。此外,與圖2所示的連續進氣方式中釜內溫度能快速達到最大值不同,圖7中釜內溫度隨時間的變化呈現升高、穩定、再升高、降低的變化趨勢。可將一次性進氣下體系的釜內溫度升高分為兩段:通入甲烷氣后便出現第一次溫升(升高約3.0℃);70min之后出現第二次溫升(升高約5.5℃)。實驗中測得一次性進氣體系中形成水合物所消耗的甲烷氣體量為 2.50L,是約為連續進氣體系中甲烷氣體消耗量的一半,說明一次性進氣條件下形成的水合物量較少。對比圖2和圖7可知,在初始溫度和實驗壓力相同的條件下,連續進氣方式比一次性進氣方式更有利于提高體系的Tmax和ΔTmax值。

圖7 初始溫度為4℃,一次性進氣下體系的釜內溫度和壓力變化圖
2.3.2 間歇進氣方式
圖8與圖9分別給出了間隔時間為10min和30min的間歇進氣方式下釜內溫度和壓力變化圖。在圖8與圖9中,如S段所示,釜壓未達實驗壓力10MPa前,釜壓按照梯狀方式增加,釜內溫度也呈現了類似的增加趨勢。當釜壓達到10MPa后,釜內溫度急劇上升且迅速達到各自的 Tmax值 20.4℃和20.5℃。這是因為在釜壓達 10MPa時,釜內溫度約為6.5℃,而6.5℃對應的環戊烷-甲烷水合物相平衡壓力略小于 0.16MPa[19,23],此時釜內壓力驅動力較大,并且S段中有5次較大的增壓擾動,兩個因素均有利于釜內水合物的快速生成,因此,釜內溫度上升快速且幅度較大。然而,隨著釜內溫度不斷升高,當釜溫達Tmax后,反應釜內壓力驅動力減小,水合物生成速率降低,水合物生成過程中釋放的相變熱熱量有限,同時受體系向低溫環境散熱的影響,釜內溫度不再升高并呈現出下降的趨勢。

圖8 初始溫度為4℃、間隔時間為10min的間歇進氣下體系的釜內溫度和壓力變化圖

圖9 初始溫度為4℃、間隔時間為30min的間歇進氣下體系的釜內溫度和壓力變化圖

表1 不同間隔時間間歇進氣方式下的實驗數據表
表1為不同間隔時間間歇進氣方式下的實驗數據表,其中,定義開始注氣到釜內溫度達最大值所用的時間為表觀溫升時間t1,Δti為間歇進氣的間隔時間。對比圖2、圖8與圖9可知,t1依次增加,主要與間隔時間Δti有關。將 t1與總間隔時間 4Δti之差定義為實際溫升時間t2,由表1可知t2數值相近。t3為反應釜內Tmax維持穩定的時間,由表1可知t3隨間隔時間增大而增加。與表1對應的不同間隔時間的間歇進氣方式下釜內溫度變化曲線如圖10所示。由圖10和表1可知,間隔時間為0min、 10min和30min下的Tmax數值分別為18.8℃、20.4℃和 20.5℃,對應的 t3則分別為45min、116min和143min。可見間歇進氣方式比連續進氣方式下的Tmax與t3值均要大。間歇進氣方式中間隔時間的長短對Tmax數值的影響較小,但對t3的大小影響顯著。較長的間隔時間對應著更大的t3值,t3值越大意味著Tmax維持的時間越長,即釜內維持高溫段的時間越長,這種釜內溫度變化特性有利于水合物生成熱熱量的有效利用。同時,由表1中可知,間歇進氣方式比連續進氣方式消耗的甲烷氣體量 Vm更大,在達到實驗壓力前加入適當的壓力擾動有利于增加水合物的總生成量。綜合可知,間歇進氣方式能達到比連續進氣方式更大的 Tmax值和ΔTmax值,并且較大的間隔時間能較大程度地提高Tmax維持穩定的時間t3。因此,間歇進氣方式為一種較優的進氣方式。

圖10 初始溫度為4℃,不同間隔時間間歇進氣下體系的釜內溫度變化圖
3 熱量分析
本實驗開始前,氣體、液體及反應裝置均處于控制溫度為T0條件下的低溫室中。因反應釜保溫層的保溫性能有限,體系在整個實驗過程中存在一定的熱量損耗。其中,體系熱量的損耗包括體系熱量向鋼釜層和保溫層的轉移以及向低溫環境的散失。在實驗過程中,釋放的水合物生成熱部分用來加熱釜內水合物漿與自由甲烷氣,其余的熱量轉移到鋼釜層和保溫層以及低溫環境中。反應持續的時間越長,體系向周圍低溫環境散失的熱量就越多。因此,釜內最高溫度 Tmax和釜內最大溫升ΔTmax受水合生成速率和體系散熱速率兩方面的影響。
釜內環戊烷-甲烷水合物生成熱的熱量去向具體表現在以下5個方面:①釜內反應液和水合物因溫度升高而吸收的熱量Qh,w,c;②釜內自由甲烷氣因溫升而吸收的熱量Qm;③316不銹鋼釜溫度升高吸收的熱量Qs;④保溫層溫度升高吸收的熱量Qa;⑤體系向低溫環境散失的熱量Qd。另外,通入釜內的甲烷氣溶解于反應液中會釋放一定量的溶解熱,但由于該溶解熱遠小于反應過程中釜內釋放的水合物生成熱,以下計算忽略溶解熱部分,并假設5個方面的熱量全部來自于反應釜內水合物生成熱的總熱量Qf,即有Qf=Qh,w,c+Qm+Qs+Qa+Qd。并將有效熱量Qh,w,c與Qf的比值定義為熱量有效利用率,其表達式如式(1)。

以圖2條件下水合物的生成過程為例,定義體系溫降較明顯處為實驗反應結束點,詳細分析該過程中釋放的水合物生成熱熱量在整個裝置中的具體分布情況。該條件下,生成的水合物所消耗的甲烷氣的體積為 5.66L,由狀態方程 PV = ZnRT得 n = 0.236mol,其中T為室溫20℃。假設本實驗中最終生成的水合物全部為環戊烷-甲烷水合物,并且分子構成全部為理想分子式8CP·16CH4·136H2·O,則水合物晶體結構中含有的環戊烷與水的總量分別為0.118mol和2.006mol。
環戊烷-甲烷水合物釋放的總生成熱熱量如式(2)。

純水體系中環戊烷-甲烷水合物生成熱在溫度范圍286~292K間變化較小,此時qf為129~130 kJ/mol[18-19],此處取為 129.5kJ/mol,則由式(1)得Qf=30.56kJ。
對于Qh,w,c的計算,公式為式(3)。
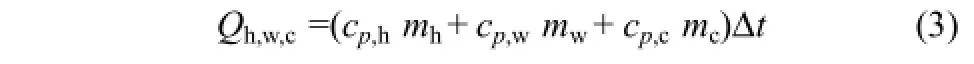
式中,cp,h、cp,w和cp,c分別環戊烷-甲烷水合物、水和環戊烷的定壓比熱容,近似取cp,h=2.8J/(g·K)[1,26]、cp,w=4.2J/(g·K)、cp,c=1.8J/(g·K);mh、mw和mc分別環戊烷-甲烷水合物、水和環戊烷的質量,計算得mh=48.1g、mw=17.4g、mc=4.0g;Δt為釜內反應液和水合物的最大溫升值,Δt=14.8℃。
對于Qm的計算,公式為式(4)。

式中,cp,m為甲烷氣體的定壓比熱容,取cp,m= 3.1J/(g·K);mm為甲烷氣體的質量,計算得 mm= 4.27g;Δt為釜中甲烷氣的最大溫升值,Δt=14.8℃。
對于Qs的計算,公式為式(5)。
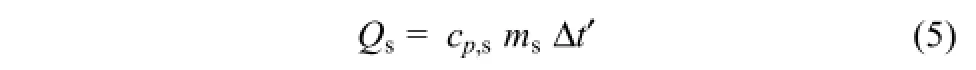
式中,cp,s為316不銹鋼定壓比熱容,取cp,s為0.50J/(g·K);ms為 316不銹鋼的質量,計算得 ms=520.2g;Δt′為鋼釜的平均溫升,因鋼釜熱導率和熱擴散系數均較大,鋼釜厚度薄,側壁厚2.5mm,底部厚10mm,熱阻Rs小,緊鄰鋼釜層外包裹有保溫層,體系向低溫環境散熱熱流量Φ 也較小,鋼層ΔT=RsΦ 較小,可認為鋼釜整體處于均一溫度,此處Δt′近似取值為比Δt低0.8℃,即Δt′=14.0℃。
對于Qa的計算,公式為式(6)。
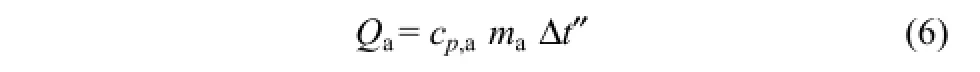
式中,cp,a為氣凝膠定壓比熱容,取 cp,a=0.56 J/(g·K)[27];ma為氣凝膠的質量,計算得ma=662.5g;Δt′為保溫層的平均溫升,對于保溫層,散熱熱流量Φ 雖小,但熱阻Rs較大,因保溫層包裹圓柱型反應釜,為空心圓柱型結構,其溫度分布復雜,將保溫層的平均溫度采用等效溫度簡化計算,此處Δt″近似取值為比Δt′低6℃,即Δt″=8.0℃。
對于Qd的計算,因直接計算體系向低溫環境的散熱量較為復雜,此處采用間接計算方式,如式(7)。

各熱量具體計算數值見表2。表2中列出了實驗過程中水合物生成熱在不同初始溫度(4℃、8℃和12℃)下的各熱量具體數值和各熱量占水合物生成熱總熱量的百分比及各熱量有效利用率。

表2 反應體系各熱量計算數值及百分比
由表2可知,3種初始溫度下的熱量有效利用率η在10.41%~14.84%之間,說明水合物生成熱總熱量的有效利用率有限。僅有少于15%的熱量用于釜內液體的加熱,而絕大部分熱量(占總熱量的比例高達85%以上)被反應裝置自身吸收和釜體向低溫環境擴散。其中,釜體向環境擴散的熱量占52.85%~67.25%,為生成熱熱量損失的最主要去向。由于實驗前體系整體與低溫環境溫度相同,此時鋼釜層與保溫層在實驗過程中吸收的熱量無法避免。從表2可知,在不同初始溫度下,Qh,w,c、Qs、Qa三者的值相近,但明顯小于Qd。初始溫度增加,Qd占總熱量的比例減小,而 Qh,w,c、Qs、Qa的比例在增大,熱量有效利用率η也增大,可見提高初始溫度可減少體系熱量向低溫環境的擴散。由以上熱量分析可知,提高反應釜的絕熱性能,能有效地增大水合物生成熱的熱量有效利用率,并使 Tmax和ΔTmax均有一定程度的增加。
4 結 論
本文進行了環戊烷-甲烷水合物生成過程溫度特性的實驗研究,考察了初始溫度、壓力和進氣方式對釜內溫度的影響,并對體系中水合物生成熱的熱量去向進行了分析,得出了以下幾點結論。
(1)初始溫度越低,Tmax越低,ΔTmax越高。
(2)壓力越高,Tmax越高,ΔTmax也越高。
(3)相對于連續進氣和一次性進氣方式,間歇進氣方式可達到更大的Tmax和ΔTmax。
(4) 在初始溫度為4℃、壓力為10MPa和間斷進氣時間為 30min的反應條件下,ΔTmax可達16.5℃。
(5)實驗過程中,較小一部分熱量留在反應釜內,絕大部分熱量會向低溫環境擴散,提高裝置的絕熱性能可使Tmax和ΔTmax均有一定程度的增加。
符 號 說 明
cp——定壓比熱容,J/(g·K)
H——氣體水合物的相變熱,kJ/mol
m——質量,g
n——物質的量,mol
Qh,w,c——釜內反應液和水合物因溫度升高而吸收的熱量,kJ
Qm——釜內自由甲烷氣因溫升而吸收的熱量,kJ
Qs——316不銹鋼釜溫度升高吸收的熱量,kJ
Qa——保溫層溫度升高吸收的熱量,kJ
Qd——體系向低溫環境散失的熱量,kJ
Qf——水合物生成熱的總熱量,kJ
qf——單位物質的量的水合物生成熱熱量,kJ
R——通用氣體常數,8.314 J/(mol·K)
Ti——釜內溫度(i=1,2,3),℃
T0——反應初始溫度,℃
Te——低溫室溫度,℃
Tmax——釜內最高溫度,℃
ΔTmax——釜內最大溫升,℃
t1——表觀溫升時間,min
t2——實際溫升時間,min
t3——反應釜內釜內最高溫度維持穩定的時間,min
Δt——釜內溫升,℃
Δti——間歇進氣的間隔時間,min
Δt′——316不銹鋼釜層的溫升,℃
Δt″——氣凝膠層的溫升,℃
Vm——甲烷氣體的消耗量,L
Z——壓縮因子
λ——熱導率,W/(m·K)
η——熱量有效利用率
下角標
a—— 環戊烷-甲烷水合物
c——水
h——環戊烷
m——甲烷
s——316不銹鋼
w——氣凝膠
參 考 文 獻
[1] SLOAN E D,KOH C A. Clathrate hydrates of natural gases[M]. Florida,US:CRC Press,2007.
[2] 任宏波,相鳳奎,張磊,等. 水合物法海水淡化技術應用進展[J]海洋地質前沿,2011(6):74-78.
[3] GHALAVAND Y,HATAMIPOUR M S,RAHIMI A. A review on energy consumption of desalination processes[J]. Desalination and
[4] LINGA P,ADEYEMO A,ENGLEZOS P. Medium-pressure Water Treatment,2015,54(6):1526-1541. clathrate hydrate/membrane hybrid process for postcombustion capture of carbon dioxide[J]. Environmental Science &
[5] LI X S,XU C G,CHEN Z Y,et al. Hydrate-based pre-combustion Technology,2007,42(1):315-320. carbon dioxide capture process in the system with tetra-n-butyl
[6] BI Y H,GUO T M, ZHU T G,et al. Influence of volumetric-flow ammonium bromide solution in the presence of cyclopentane[J]. Energy,2011,36(3):1394-1403. rate in the crystallizer on the gas-hydrate cool-storage process in a new gas-hydrate cool-storage system[J]. Applied Energy,2004,78
[7] 焦麗君,孫志高,趙之貴,等. 添加劑對水合物蓄冷過程影響探討[J]. 科學技術與工程,2014,14(32):217-220.
[8] AMAN Z M,OLCOTT K,PFEIFFER K,et al. surfactant adsorption and interfacial tension investigations on cyclopentane hydrate[J]. Langmuir,2013,29(8):2676-2682. (1):111-121.
[9] LV Q N,LI X S,XU C G,et al. Experimental investigation of the formation of cyclopentane-methane hydrate in a novel and large-size bubble column reactor[J]. Industrial & Engineering Chemistry Research,2012,51(17):5967-5975.
[10] OYAMA H,SHIMADA W,EBINUMA T,et al. Phase diagram,latent heat,and specific heat of TBAB semiclathrate hydrate crystals[J]. Fluid Phase Equilibria,2005,234(1):131-135.
[11] XU C G,ZHANG S H,CAI J,et al. CO2(carbon dioxide) separation from CO2-H2(hydrogen) gas mixtures by gas hydrates in TBAB (tetra-n-butyl ammonium bromide) solution and Raman spectroscopic analysis[J]. Energy,2013,59:719-725.
[12] STROBEL T A,TAYLOR C J,HESTER K C,et al. Molecular hydrogen storage in binary THF-H2clathrate hydrates[J]. The Journal of Physical Chemistry B,2006,110(34):17121-17125.
[13] LI X S,CAI J,CHEN Z Y,et al. Hydrate-based methane separation from the drainage coal-bed methane with tetrahydrofuran solution in the presence of sodium dodecyl sulfate[J]. Energy & Fuels,2012,
[14] SUN Z G,WANG R Z,MA R S,et al. Natural gas storage in 26(2):1144-1151. hydrates with the presence of promoters[J]. Energy Conversion and
[15] 梁德青,郭開華,樊栓獅,等. HCFC-141b氣體水合物融解熱的DSC測試[J]. 工程熱物理學報,2002(s1):47-49. Management,2003,44(17):2733-2742.
[16] ZHANG Y,DEBENEDETTI P G,PRUD'HOMM R K,et al. Differential scanning calorimetry studies of clathrate hydrate
[17] GUPTA A,LACHANCE J,SLOAN E D,et al.Measurements of formation[J].The Journal of Physical Chemistry B,2004,108(43): 16717-16722. methane hydrate heat of dissociation using high pressure differential scanning calorimetry[J]. Chemical Engineering Science,2008,63 (24):5848-5853.
[18] CHEN Z Y,LI Q P,YAN Z Y,et al.Phase equilibrium and dissociation enthalpies for cyclopentane+ methane hydrates in NaCl aqueous solutions [J]. The Journal of Chemical & Engineering Data,
[19] LV Q N,LI X S,CHEN Z Y,et al. Phase equilibrium and 2010,55(10):4444-4449. dissociation enthalpies for hydrates of various water-insoluble organic promoters with methane[J]. Journal of Chemical & Engineering Data,2013,58(11):3249-3253.
[20] 陳朝陽,李小森,顏克鳳,等. 一種開采天然氣水合物的方法及裝置:101016841A[P]. 2007-02-13.
[21] CHEN Z Y,FENG J C,LI X S,et al. Preparation of warm brine in 2014,53(36):14142-14157.
[22] 閆忠元,陳朝陽,李小森,等. 鹽水體系中環戊烷-甲烷水合物相平衡測定與模擬[J]. 過程工程學報,2010(3):476-481. situ seafloor based on the hydrate process for marine gas hydrate thermal stimulation[J]. Industrial & Engineering Chemistry Research,
[23] SUN Z G,FAN S S,GUO K H,et al.Gas hydrate phase equilibrium data of cyclohexane and cyclopentane[J]. Journal of Chemical & Engineering Data, 2002,47(2):313-315.
[24] 孫長宇,黃強,陳光進. 氣體水合物形成的熱力學與動力學研究進展[J]. 化工學報,2006,67(5):1031-1039.
[25] FAN S S,LIANG D Q,GUO K H.Hydrate equilibrium conditions for cyclopentane and a quaternary cyclopentane-rich mixture[J]. Journal of Chemical & Engineering Data,2001,46(4):930-932.
[26] TOMBARI E,PRESTO S,SALVETTI G,et al. Heat capacity of tetrahydrofuran clathrate hydrate and of its components,and the clathrate formation from supercooled melt[J]. The Journal of Chemical physics,2006,124(15):154507.
[27] 周祥發,馮堅,肖漢寧,等. 二氧化硅氣凝膠隔熱復合材料的性能及其瞬態傳熱模擬[J]. 國防科技大學學報,2009,31(2): 36-40,69.
System temperature properties in the process of the cyclopentane-methane binary hydrates formation
HU Yafei1,2,CAI Jing1,LI Xiaosen1
(1Guangdong Key Laboratory of New and Renewable Energy Resenrch and Development,Key Laboratory of Gas Hydrate,CAS,Guangzhou Institute of Energy Conversion,Chinese Academy of Sciences,Guangzhou 510640,Guangdong,China;2University of Chinese Academy of Sciences,Beijing 100049,China)
Abstract:In this paper,the changes of the system temperatures and heat loss were investigated during the formation of cyclopentane-methane binary hydrates. The system temperature measurements were carried out under the conditions of initial temperatures of 4℃,8℃,and12℃,pressures of 2MPa,4MPa,6MPa,8MPa and 10MPa,and different gas injection modes (single,continuous and intermittent). The maximum temperature (Tmax) and the maximum temperature increase (ΔTmax) in the system were compared. The experimental results illustrate that the pressures and gas injection modes have significant influence on Tmaxwhile the initial temperatures,pressures and gas injection modes all significantly effect ΔTmax. Thus,the conditions of lower initial temperature,higher pressure and injecting intermittently help to increase ΔTmax. Under the condition of 4℃ and 10MPa,intermittent injection with the interval time of 30minute,the maximum value of ΔTmaxis 16.5℃. In addition,the heat analysis results indicate that the main heat loss is from the inner reactor to the outside cold environment. Therefore,improving the insulation properties of insulating layer is helpful to enhancethe heating efficiency in the process of the cyclopentane-methane binary hydrates formation.
Key words:hydrate;methane;cyclopentane;formation heat;maximum temperature increase degree
中圖分類號:TQ 026
文獻標志碼:A
文章編號:1000-6613(2016)05-1418-10
DOI:10.16085/j.issn.1000-6613.2016.05.022
收稿日期:2015-09-28;修改稿日期:2015-11-02。
基金項目:國家杰出青年科學基金(51225603)、國家自然科學基金(51376184)、中石油-中科院高端戰略聯盟計劃(2015A-4813-2)及中海油研究總院委托項目(CRI2015RCPS0053OCN)。
