辛蒼顆粒中藥材的定性鑒別和黃芩苷的含量測定*
2016-06-27 07:37:17陳華于波濤范開華金偉華蒲志強王詩華謝秋
西部醫學 2016年6期
陳華 于波濤 范開華 金偉華 蒲志強 王詩華 謝秋
(解放軍成都軍區總醫院藥劑科, 四川 成都 610083)
·論著·
辛蒼顆粒中藥材的定性鑒別和黃芩苷的含量測定*
陳華于波濤范開華金偉華蒲志強王詩華謝秋
(解放軍成都軍區總醫院藥劑科, 四川 成都 610083)
【摘要】目的建立辛蒼顆粒質量控制標準。方法采用薄層色譜法(TLC)對辛蒼顆粒處方中的黃芩、甘草、黃芪進行定性鑒別;采用高效液相色譜法(HPLC)對處方中的黃芩苷進行含量測定,色譜柱為Agilent HC-C18(4.6mm ×250mm,5μm),流動相為甲醇-0.1%磷酸(40∶60),流速為1.0ml/min,檢測波長為280nm,柱溫為34℃。結果TLC斑點清晰,分離度較好,專屬性強,陰性對照無干擾;黃芩苷在1.92~61.44μg/ml范圍內濃度與峰面積呈良好的線性關系(r=0.9999),平均回收率為100.30%,RSD=1.82%(n=9)。結論該研究方法準確可靠,重復性好,且簡便可行,可用于辛蒼顆粒的質量控制。
【關鍵詞】辛蒼顆粒;質量標準;黃芩苷;薄層色譜法;高效液相色譜法
辛蒼顆粒是由白芷、川芎、蒼耳子、黃芪、梔子等11味中藥材經提取精制而成的醫院制劑,批準文號為成制字(2011)F02010號,具有清熱、解毒、祛風、通竅等功效,主治鼻塞、流鼻涕、感冒等。經過多年的臨床應用,已成為療效確切的驗方,原質量控制標準方法較為簡單。為適應國家對醫療機構制劑室進行GPP管理和軍隊醫療機構制劑標準提高計劃要求,有效控制該制劑質量標準,保證臨床用藥安全有效,本研究結合現有技術條件,采用薄層色譜法(TLC)對處方中黃芩、甘草、黃芪進行定性鑒別,采用高效液相色譜法(HPLC)對處方中黃芩苷含量進行測定,現將結果報告如下。
1儀器與試藥
1.1儀器HP 1100高效液相色譜儀[G1311A Quat Pump泵,G1315A DAD 紫外檢測器,Agilent HC-C18(4.6 mm×250 mm,5μm)分析柱,美國Agilent公司];AB135-S型精密電子天平(梅特勒-托利多儀器公司,精度:0.01mg);AE-200S型電子天秤(梅特勒-托利多儀器公司,精度:0.1mg);VGT-1990QTD型超聲儀(蘇州江東精密儀器有限公司);TGL-18G-C型高速臺式離心機(上海安亭科學儀器廠);DU730型紫外分光光度計(美國貝克曼儀器公司);HHS11-Ni2電熱恒溫水浴鍋(北京長安永創科學儀器有限公司);SZ-97型自動三重純水蒸餾器(上海亞榮生化儀器廠)。
1.2藥品與試劑辛蒼顆粒樣品(成都軍區總醫院,規格:5g/袋,批號:150120、150121、150122),甘草對照藥材(批號:120904-201318),黃芪對照藥材(批號:121462-201304),均由中國食品藥品檢定研究院提供;黃芩苷對照品(批號:110715-201117,純度:97.0%)由中國食品藥品檢定研究院提供;陰性樣品均為自制;甲醇為色譜純,其它試劑均為分析純;水為三重蒸餾水;硅膠G薄層板(青島海洋化工廠)。
2方法與結果
2.1 TLC鑒別
2.1.1黃芩的TLC鑒別[2,3]取供試品10g,研細,加甲醇30ml,加熱回流30min,濾過,濾液蒸干,殘渣加甲醇2ml溶解,離心,取上清液,作為供試品溶液。另取黃芩苷對照品適量,加甲醇制成1.0mg/ml的照品溶液。取缺黃芩陰性樣品10g,同供試品溶液制備方法制成陰性對照溶液。參照2010年版《中國藥典》一部中TLC法[1],分別吸取黃芩苷對照品溶液2μl,供試品溶液和陰性對照品溶液各3μl,點于同一含4%醋酸鈉的羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以乙酸乙酯-丁酮-甲酸-水(5∶3∶1∶1)為展開劑,預飽和15min,展開,取出,晾干,噴以1%三氯化鐵乙醇溶液,晾干至斑點顯色清晰。結果顯示,在供試品色譜中,與對照品色譜相應位置上顯相同顏色的斑點,陰性對照無干擾,見圖1。
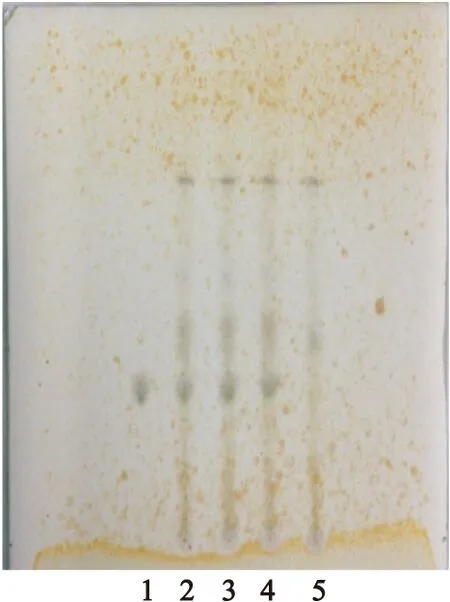
圖1黃芩TLC圖
Figure 1TLC of Radix Scutellariae
注:1.黃芩苷對照品;2~4.供試品;5.陰性對照
2.1.2甘草的TLC鑒別[4,5]取供試品10g,研細,加三氯甲烷25ml,加熱回流1小時,濾過,棄去三氯甲烷液,藥渣揮干溶劑,加水5ml攪拌濕潤,加水飽和正丁醇50ml,超聲處理30min,濾過,取正丁醇液蒸干,殘渣加甲醇2ml使溶解,作為供試品溶液。另取甘草對照藥材1g和缺甘草的陰性樣品10g,同法分別制成對照藥材溶液和陰性對照溶液。參照2010年版《中國藥典》一部中TLC法[1],分別吸取對照藥材溶液6μl、供試品溶液和陰性對照溶液各15μl,點于同一硅膠G薄層板上,以乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)為展開劑,預飽和15min,展開,取出,晾干,噴以10%硫酸乙醇溶液,在105℃加熱至斑點顯色清晰并置紫外光燈(365nm)下檢視。結果顯示,在供試品色譜中,與對照藥材色譜相應位置上顯相同顏色的斑點,陰性對照無干擾,見圖2。
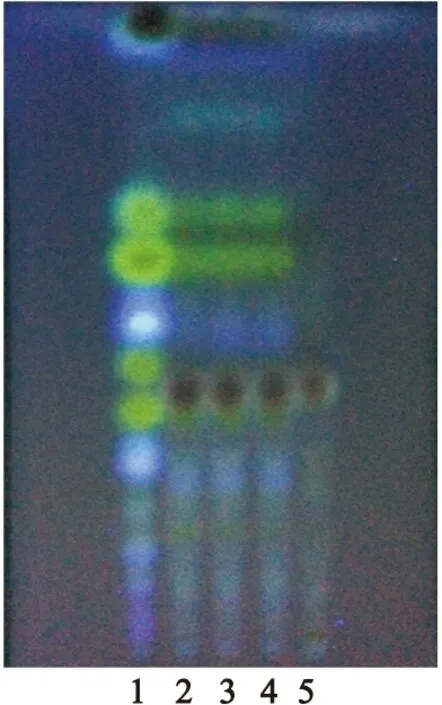
圖2甘草TLC圖
Figure 2TLC of Radix et Rhizoma Glycyrrhizae
注:1.甘草對照藥材;2~4.供試品;5.陰性對照
2.1.3黃芪的TLC鑒別[6,7]取供試品10g,研細,加甲醇10ml,超聲處理30min,濾過,濾液蒸干,殘渣加水20ml溶解,用水飽和正丁醇振搖提取2次,每次20ml,合并正丁醇液,用氨試液洗滌2次,每次20ml,棄去氨試液,正丁醇液蒸干,殘渣加甲醇1ml使溶解,作為供試品溶液。另取黃芪對照藥材1g和缺黃芪陰性樣品10g,同法制成對照藥材溶液和陰性對照品溶液。參照2010年版《中國藥典》一部中TLC法[1],分別吸取對照藥材溶液8μl、供試品溶液和陰性對照溶液10μl,點于同一硅膠G薄層板上,以乙酸乙酯-丁酮-甲酸-水(5∶3∶1∶1)為展開劑,預飽和15min,展開,取出,晾干,噴以10%硫酸乙醇溶液,在105℃加熱至斑點顯色清晰后置紫外光燈(365nm) 下檢視。結果顯示,在供試品色譜中,在與對照藥材色譜相應位置上顯相同顏色的斑點,陰性對照無干擾,見圖3。
2.2黃芩苷的含量測定[8-10]
2.2.1色譜條件與系統適用性試驗 色譜柱:Agilent HC-C18(4.6mm×250mm,5 μm);流動相:甲醇-0.1%磷酸(40∶60);流速:1ml/min;檢測波長:280nm;柱溫:34℃;進樣量:10μl,理論板數按黃芩苷峰計算應不低于3000。
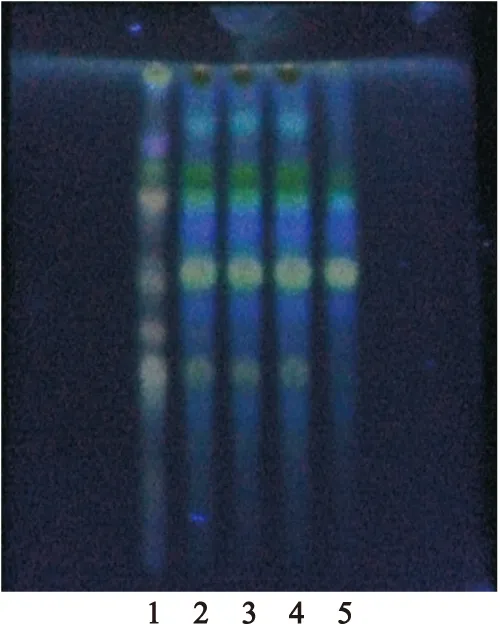
圖3黃芪TLC圖
Figure 3TLC of Astragali Radix
注:1.黃芪對照藥材;2~4.供試品;5.陰性對照
2.2.2對照品溶液的制備精密稱取干燥至恒重的黃芩苷對照品適量,加甲醇制成50μg/ml的溶液,即得。
2.2.3供試品溶液的制備取裝量差異下的供試品適量,研細,取約0.5g,精密稱定,置具塞錐形瓶中,精密加入50%甲醇75 ml,密塞,稱定重量,超聲處理20min,放冷,稱定重量,用50%甲醇補足減失的重量,搖勻,離心,濾過,取續濾液,即得。
2.2.4陰性對照溶液的制備取缺黃芩的陰性樣品適量,按“2.2.3”項下方法制備陰性對照溶液,即得。
2.2.5專屬性試驗精密吸取上述黃芩苷對照品溶液、供試品溶液、陰性對照溶液各10μl, 按上述色譜條件進樣測定,記錄色譜圖。結果顯示,陰性對照溶液在與黃芩苷對照品溶液相應的出峰處未見吸收峰,表明陰性對照溶液無干擾,見圖4。
2.2.6線性關系考察精密稱取黃芩苷對照品適量,加甲醇溶液制成76.8μg/ml的對照品儲備溶液。再分別取適量對照品儲備溶液,精密量取,加甲醇適量,分別制成每1ml含1.92、3.84、7.68、15.36、30.72、61.44μg的對照品溶液。分別精密吸取上述對照品溶液10μl,按上述色譜條件進樣測定,記錄色譜圖。以對照品的測定濃度(x,mg/ml)為橫坐標,峰面積積分值(y)為縱坐標,進行線性回歸,得回歸方程為:y=23.939x-5.5149(r=0.9999)。結果顯示,黃芩苷在1.92~61.44μg范圍內濃度與峰面積積分值呈良好的線性關系。
2.2.7精密度試驗精密吸取 “2.2.6”項下61.44μg/ml黃芩苷對照品溶液10μl,注入液相色譜儀,按上述色譜條件測定,連續進樣6次,記錄色譜圖。結果顯示,計算RSD=0.16%(n=6),表明儀器精密度良好。
圖4高效液相色譜圖
Figure 4HPLC chromatograms
注:A. 黃芩苷對照品;B. 供試液品;C. 陰性對照品;1.黃芩苷
2.2.8穩定性試驗取同一批樣品,按“2.2.3”項下方法制備供試品溶液,在室溫下放置,分別于0、2、4、8、12,24小時各進樣1次,按上述色譜條件測定,記錄峰面積。結果顯示, RSD=0.54%(n=6),表明供試品溶液在24小時內穩定性良好。
2.2.9重復性試驗分別取同一批號樣品6份約0.5g,精密稱定,按“2.2.3”項下方法制備供試品溶液,進樣6次,按上述色譜條件測定,記錄峰面積。結果顯示, RSD=1.0%(n=6),表明本法重復性良好。
2.2.10加樣回收率試驗取已知含量的樣品9份,約0.25g,精密稱定,按3份為1組,每組中分別按樣品中黃芩苷含量的80%、100%、120%精密加入對照品溶液,按照“2.2.3”項下方法制備供試液,進樣9次,按上述色譜條件測定,計算加樣回收率,見表1。
2.2.11樣品含量測定取3批供試品各適量,分別按 “2.2.3”項下方法制備供試品溶液,按上述色譜條件測定,記錄峰面積,計算樣品中黃芩苷的含量,見表2。
3討論
辛蒼顆粒為中藥復方制劑,成分較多,彼此干擾,在供試品的前處理過程中,通過提取、分離、純化,使溶液成分由復雜變簡單,減少干擾。在甘草TLC鑒別中,以三氯甲烷為提取溶劑,提取后棄去三氯甲烷液,可減少脂溶性成分干擾。在黃芩TLC鑒別時,參考《中國藥典》2010版一部方法,黃芩鑒別使用的薄層板為聚酰胺薄膜,經實驗后修訂采用含4%醋酸鈉的羧甲基纖維素鈉為黏合劑的硅膠G薄層板,系臨用前自制的薄層板;展開劑篩選過程中,以乙酸乙酯-丁酮-甲酸-水(5∶3∶1∶1)為展開劑,展開后斑點清晰易觀察,Rf值適中,斑點清晰勻稱,陰性無干擾,重現性好。
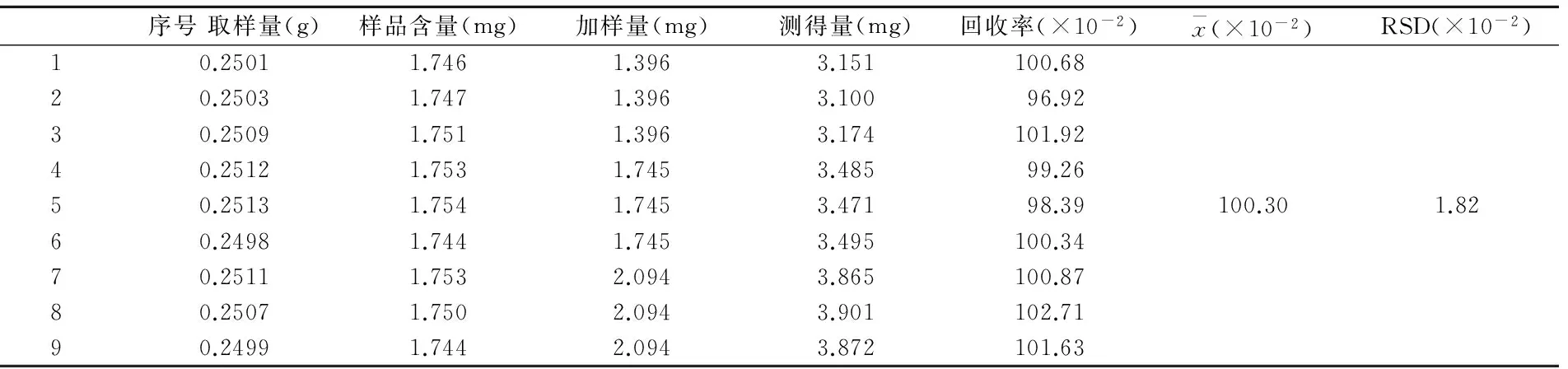
表1 加樣回收率試驗結果(n=9)
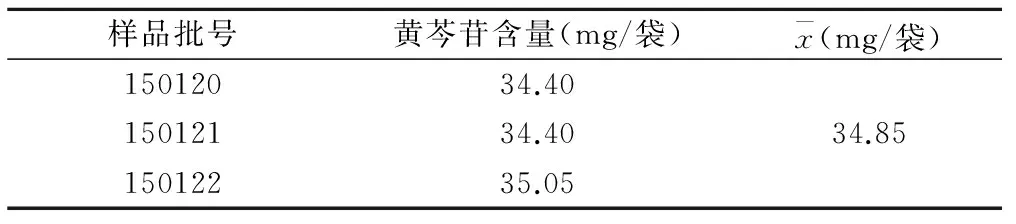
表2 樣品中黃芩苷的含量
在黃芩苷含量測定項下流動相的選擇中, 參照《中國藥典》2010 年版及有關文獻,試用多種流動相體系, 甲醇-水-磷酸 (47∶53∶0.2) 、甲醇-0.1%磷酸 (40∶60) 、甲醇-0.3%磷酸 (20∶80)、 甲醇-冰醋酸-水 (40∶1∶60)進行試驗, 結果表明,用甲醇-0.1%磷酸溶液 (40∶60)作為流動相,樣品分離度較好,峰型佳,保留時間穩定,無雜質峰干擾。
目前,《中國人民解放軍醫療機構制劑規范》(以下簡稱《軍規》)雖然經歷了多次修訂,但隨著醫藥檢驗技術的發展和安全監管要求的提高,與現行版《中國藥典》等國家藥品標準相比差距較大,突出表現在以下幾個方面。①部分制劑處方工藝不合理。②檢驗項目設置不夠齊全。③檢測方法落后,當前通用的技術和設備應用較少。④質控指標偏低,難以控制制劑質量。⑤凡例通則項明顯落后于現版《中國藥典》,難以保證藥品安全、有效和質量可控。⑥非標準制劑審批把握尺度寬嚴不一,批準后未再進行系統評價和驗證,其執行標準中存在的問題比標準制劑更為嚴重[11]。軍隊醫療機構制劑標準提高計劃是以確保人民群眾與官兵用藥安全為根本目的,以提高藥品標準和藥品質量為工作重心,完善監管體制,創新監管機制,依法科學實施監管。通過本次制劑標準提高計劃,能夠對制劑質量標準有個整體的推進,并初步建立制劑的準入和退出標準,同時完善標準提高的長效機制,將加快《軍規》向《中國藥典》等國家標準看齊的步伐,更加完善軍隊醫療制劑的監管制度,全面保障軍民用藥安全有效。
此次啟動“軍隊制劑標準提高計劃”對軍隊醫療機構制劑的發展是一種強有力的引導,尤其會對軍隊醫療機構制劑滑坡現狀起到推動作用[12],引導配制單位爭取各種資金,加大研發投入,引導醫院制劑向著重視研發的良性道路發展。
4結論
本研究建立的辛蒼顆粒定性鑒別方法,特征斑點明顯,分離度佳,陰性對照無干擾;建立的定量方法,精密度、重復性、穩定性均符合要求,所建標準可用于辛蒼顆粒質量控制。
【參考文獻】
[1]國家藥典委員會.中華人民共和國藥典一部[S]. 2010年版.北京:中國醫藥科技出版社,2010 :282.
[2]付延偉,紀松崗,李明春,等.復方茵陳顆粒的質量標準研究[J].解放軍藥學學報,2015,31(1):53-56.
[3]胡北,馬宏達,張朝紳,等. 和肝利膽顆粒的質量標準研究[J].中國藥房,2014,25(39):3679-3681.
[4]王輝,袁海銘,陳梅榮.羚羊感冒片薄層臨別研究[J].江西中醫藥,2008,39(2):45-46.
[5]黃可婧,王麗娟.附子理中片的質量標準研究[J].天津藥學,2014,26(5):16-20.
[6]農毅清,蔣林,譚安薔.癌痛消顆粒質量標準研究[J].中國藥師,2014,17(11):1859-1832.
[7]尹華,章建華,張春霞,等.芪參健骨顆粒的薄層鑒別研究[J].浙江中醫藥大學學報,2011,35(1):79-81.
[8]邵紅燕,張和明.高效液相色譜法測定肩周炎痛貼中黃芩苷的含量[J].中國藥師,2009,12(10):1498-1499.
[9]馬一明,劉淑媛.高效液相色譜法測定祛斑膏中黃芩苷的含量[J],中國醫院藥學雜志, 2010,30(13):1167-1168.
[10] 邵禮梅,王云龍,李延雪.高效液相色譜法測定蒲地藍消炎片中黃芩苷與黃芩素含量[J].中國藥業,2012,21(4):37-38.
[11] 陳征宇,孔愛英. 軍隊醫療機構制劑標準提高計劃實施構想[J].中國藥事,2013,27(11):1157-1160.
[12] 文娟,張珂良,汪麗,等.對我國醫療機構制劑管理現狀的思考[J].中國藥事,2012,26 (4):321-323.
Qualitative identification of Xincang Granules and determination of Baicalin in Xincang Granules
CHEN Hua,YU Botao,FAN Kaihua,et al
(DepartmentofPharmacy,GeneralHospitalofChengduMilitaryCommand,Chengdu610083,China)
【Abstract】ObjectiveTo establish the quality standard of Xincang Granules. MethodsThe TLC method was used to qualitatively identify Radix Scutellariae, licorice and Astragali Radix. HPLC method was adopted to determine the content article of baicalin.The determination was performed on Agilent HC-C18(4.6mm ×250mm,5μm) column with mobile phase consisted on methyl alcohol-0.1% phosphoric acid(40∶60) at the flow rate of 1.0ml/min,the column temparature was 34℃ and the detection wavelength was set at 280nm.ResultsThe spots of TLC were fairly clear with the good separation. The negative samples showed no interference. The linear range of baicalin was 1.92~61.44μg/m1(r=0.9999).The average recovery rate was 100.30%. The RSD was 1.82%(n=9).ConclusionThe simple, rapid and reliable method can be used for the quality control of Xincang Granules.
【Key words】Xincang Granules;Quality standard;Baicalin; TLC;HPLC
基金項目:總后衛生部軍隊醫療機構制劑標準提高科研專項課題(14ZJZ18-2)
通信作者:范開華,主任藥師,碩士研究生導師。E-mail:fankeyi@sohu.com
【中圖分類號】R 914
【文獻標志碼】A
doi:10.3969/j.issn.1672-3511.2016.06.026
(收稿日期:2015-12-02; 編輯: 母存培)
