布洛芬-偽麻黃堿衍生物的合成
2016-03-27 01:49:35易八賢
中國藥業 2016年8期
孫 丹,呂 敏,呂 振,李 泉,易八賢
(1.黑龍江中醫藥大學,黑龍江 哈爾濱 150040; 2.哈爾濱三聯藥業股份有限公司,黑龍江 哈爾濱 150025;3.中國醫藥工業研究總院,上海 201403)
布洛芬-偽麻黃堿衍生物的合成
孫 丹1,呂 敏2,呂 振2,李 泉2,易八賢3
(1.黑龍江中醫藥大學,黑龍江 哈爾濱 150040; 2.哈爾濱三聯藥業股份有限公司,黑龍江 哈爾濱 150025;3.中國醫藥工業研究總院,上海 201403)
目的 合成、制備高純度布洛芬-偽麻黃堿衍生物,探討其理化性質及波譜特征。方法 以布洛芬和偽麻黃堿為起始原料,1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽(EDC)為縮合劑,經柱層析及重結晶純化,制備布洛芬-偽麻黃堿的酯化和酰胺衍生物,并對其結構進行檢測。結果 所得化合物純度均大于99.5%,其波譜特征與目標化合物相符。結論 該合成方法簡單易行,產物純度高,可作為雜質對照品,用于布洛芬-偽麻黃堿復方制劑的質量控制。
布洛芬;偽麻黃堿;酯化;酰胺化;雜質對照品
布洛芬(α-甲基-4-(2-甲基丙基)苯乙酸,ibuprofen)和鹽酸偽麻黃堿([S-(R*,R*)]-α-1-[1-(甲氨基)乙基]-苯甲醇鹽酸鹽,pseudoephedrine hydrochloride)的復方制劑廣泛用于治療感冒[1-2],常見的有布洛偽麻片和布洛偽麻膠囊等[3]。布洛芬化學結構中有羧基,鹽酸偽麻黃堿結構中有羥基和胺基,在復方制劑中,兩者充分接觸,經長期放置,可發生酰胺化或酯化反應,生成微量的布洛芬-偽麻黃堿酰胺或布洛芬-偽麻黃堿酯[4-6]。1-乙基-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽(EDC)是活性較高的碳二亞胺類脫水劑,是一種實用、高效的酰胺鍵、酯鍵形成試劑,且所需反應條件溫和,反應收率高,選擇性好,污染少[7-9]。本研究中選擇酰胺化反應和酯化反應的縮合劑。偽麻黃堿結構中的氨基活性強,酯化反應時需進行保護,選擇有機合成廣泛應用的叔丁氧羰基(BOC)[10-11]進行氨基保護,采用鹽酸脫保護的方法[12],同時使布洛芬 -偽麻黃堿酯成鹽酸鹽。
1 儀器與試藥
1.1 儀器
JJ-I型電動攪拌器(江蘇器械廠);DLSB-5/20型低溫冷卻液循環泵(鄭州長城科工貿有限公司);SHB-Ⅲ型循環水式真空泵(鄭州長城科工貿有限公司);RE-52AA型旋轉蒸發器(上海亞榮生化儀器廠);全自動 YRT-3型熔點儀(上海精密科學儀器有限公司);LC-2010AHT型高效液相色譜儀(日本島津公司);Inova-600型核磁共振儀(美國Varian公司);6520四極桿 -飛行時間串聯質譜儀(美國 Agilent公司);FTIR-650型傅立葉紅外光譜儀(天津港東科技發展股份有限公司)。
1.2 試藥
布洛芬(批號為C100-1303199M,純度為98.5%)和偽麻黃堿(批號為130423,純度為98.0%),由哈爾濱三聯藥業股份有限公司研發中心提供;1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽(EDC,批號為E1216007,純度為98.5%),購自上海晶純試劑有限公司;其余試劑均為市售分析純。
2 方法與結果
2.1 試驗方法
2.1.1 合成路線
布洛芬與鹽酸偽麻黃堿的分子結構見圖1。合成反應路線見圖2。
2.1.2 布洛芬-偽麻黃堿酰胺化衍生物的合成
將布洛芬13.56 g和EDC 24.97 g溶于300 mL二氯甲烷中,室溫反應30min后加入偽麻黃堿固體11.28g,反應至薄層色譜(TLC)無原料斑點。有機相水洗至水相pH為7;無水硫酸鈉干燥,濾除干燥劑,減壓濃縮后,經硅膠柱層析(石油醚/乙酸乙酯)分離,可同時得到2種白色固體,為化合物Ⅰ和化合物Ⅱ,分別經二氯甲烷/石油醚重結晶得到高純度化合物。

圖1 布洛芬和鹽酸偽麻黃堿分子結構

圖2 合成反應路線圖
2.1.3 布洛芬-偽麻黃堿酯化衍生物的合成
將偽麻黃堿固體18.06 g溶于350 mL二氯甲烷中,0℃下滴加(Boc)2O 28.76 g/二氯甲烷50 mL溶液,滴畢,室溫反應至 TLC檢測無原料斑點。有機相用1 mol/L檸檬酸、飽和食鹽水和水依次洗至pH為6,無水硫酸鎂干燥,減壓濃縮至干,得淡黃色油狀物,正己烷重結晶得白色固體,純度為 99.6%,MS m/z:288.2 [M+Na]+,553.3[2M+Na]+。
將布洛芬14.20 g和EDC 26.44 g溶于300 mL的二氯甲烷中,室溫反應30 min,加入上述固體15.50 g,反應至TLC檢測體系無原料斑點。有機相水洗,無水硫酸鈉干燥,濃縮得淡黃色油狀物,硅膠柱層析(石油醚/乙酸乙酯),得無色油狀物。
將上述油狀物用二氯甲烷120 mL溶解,于-10℃滴加氯化氫乙醇溶液32 mL,保溫反應至TLC無原料斑點,加水100 mL攪拌5 min,靜置分層,水相二氯甲烷萃取,有機相水洗,干燥,減壓濃縮得淡黃色油狀物;正己烷重結晶得白色固體,即化合物Ⅲ。
2.1.4 酰胺化及酯化衍生物純度檢測
所得化合物純度檢測參考布洛偽麻那敏復方制劑含量檢測方法[13-14],采用高效液相色譜(HPLC)法,色譜條件:色譜柱為WatersSymmetryShieldRP8柱(150mm× 4.6 mm,3.5 μm);流動相為 0.020 mol/L的 Na2HPO4(H3PO4調pH至3.5)-乙腈(50∶50);檢測波長為220nm;柱溫為25℃。
2.2 試驗結果
合成制備了3個化合物,所得化合物純度檢測結果均大于99.5%,理化性質穩定,波譜特征與目標化合物相符。詳見表1、表2和圖3。
3 討論
酯化和酰胺化衍生物結構中均包括布洛芬和偽麻黃堿2個基本構架,區別在于羧基結合位點,其理化性質及波譜特征會有所不同。化合物的1HNMR及H-H COSY數據存在明顯區別,參考質譜、紅外光譜數據分析可判斷化合物結構。
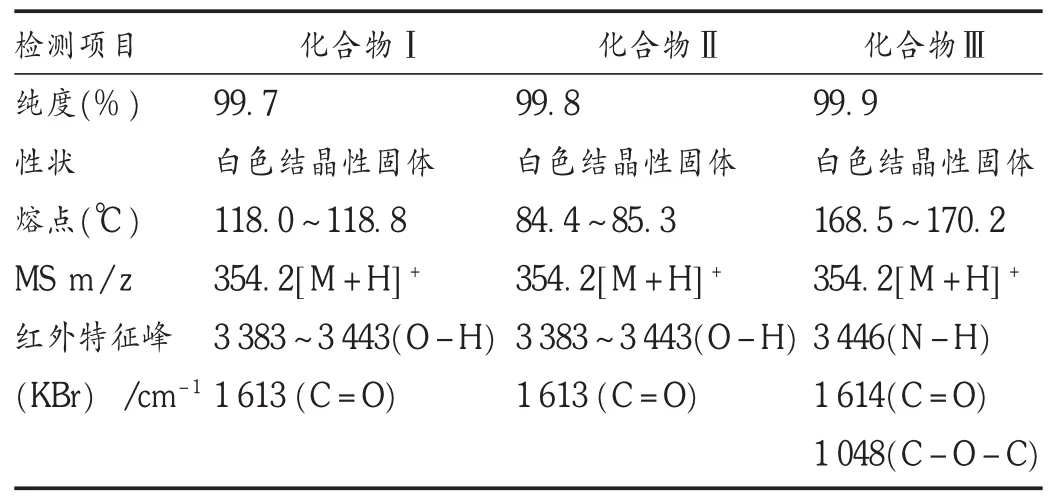
表1 化合物的理化性質、質譜及紅外光譜數據

圖3 布洛芬和偽麻黃堿結構氫編號

表2 化合物1HNMR的核磁共振氫譜數據
在化合物Ⅰ的1HNMR 中,δ0.839(6H,d,J=6.6Hz),0.869(3H,dd,J=3.0,6.6Hz),1.191(3H,d,J=7.2Hz),2.725(3H,s)處有 5個甲基氫信號;在δ2.388(2H,d,J=7.2Hz)處有 1個亞甲基氫信號;在δ1.791(1H,m),3.907(1H,q,J=6.6Hz),4.513(1H,dd,J=4.8,8.4Hz),4.705(1H,m)存在 4個次甲基信號;結合重水交換氫譜,在δ5.312(1H,d,J=4.2Hz)處為活潑氫信號;同時在方向區域存在9個芳香氫信號。在H-H COSY譜中可見δ5.312(1H,d,J=4.2 Hz)活潑氫信號與δ4.513(1H,dd,J=4.8,8.4 Hz)處的次甲基信號存在相關性。由此可推斷化合物Ⅰ以酰胺鍵的方式連接,結合質譜信號給出的準分子離子信號,紅外光譜中3 383~3 443 σ/cm-1的雙峰,符合羥基形成氫鍵后的O-H伸縮振動特征,可確定化合物結構為布洛芬-偽麻黃堿酰胺。
在化合物Ⅱ的1HNMR中,通過分析化合物Ⅱ的1HNMR及H-H COSY譜,可發現其與化合物Ⅰ存在較大相似性,其δ5.512(1H,d,J=4.2 Hz)處活潑氫信號與δ4.456(1H,dd,J=4.2,8.4 Hz)處的次甲基信號存在相關性,其紅外光譜數據與化合物Ⅰ一致。可推斷其化合物結構與化合物Ⅰ一致,即布洛芬-偽麻黃堿酰胺,與化合物Ⅰ是非對映異構體。
化合物Ⅲ中,與酯羥基相連的亞甲基氫 δ5.711(1H,d,J=9.0 Hz)的只與相鄰亞甲基氫有耦合而成雙峰,活潑氫成鹽后不再與碳上的氫有耦合,紅外光譜中3 446 σ/cm-1的為尖單峰,符合仲胺的N-H伸縮振動特征,與化合物Ⅰ、化合物Ⅱ相比,1 048 σ/cm-1的較強吸收符合酯基C—O—C的伸縮振動特征,推斷化合物Ⅲ是布洛芬-偽麻黃堿酯的鹽酸鹽。
本研究中通過化學合成得到高純度產物,通過MS,IR,1HNMR,H-H COSY等確定其結構,可作為雜質對照品,用于布洛芬-偽麻黃堿的復方制劑的質量控制。
[1]張玉潔,羅永煌,戈振凱,等.布洛芬復方制劑的研究概況[J].中國衛生產業,2011,8(8):5-6.
[2]邱 楓,孫亞欣,肇麗梅,等.布洛偽麻泡騰顆粒劑與片劑在健康人體的生物等效性[J].中國新藥與臨床雜志,2015,34(1):58-63.
[3]國家藥典委員會.中華人民共和國藥典(二部)[M].北京:中國醫藥科技出版社,2010:119.
[4]陳詩語,杭 琪,敖桂珍,等.新型布洛芬衍生物的合成及其抗炎活性[J].合成化學,2014,22(6):781-784.
[5]陳 卓,馮鎖民,楊建全,等.前藥布洛芬扁桃酸酯的合成[J].應用化工,2013,42(10):1 806-1 807.
[6]宋 妮,李英霞,孫 雪,等.布洛芬糖衍生物的合成[J].藥學學報,2004,39(2):105-109.
[7]喻 平,胡泉源,祁小云,等.EDC在有機合成中的應用研究進展[J].廣州化工,2011,39(8):9-11.
[8]Christian AGN,Falgue V.Amide bond formation and peptide coupling[J].Cheminform,2005,61(46):10 827-10 852.
[9]王哲清.實用高效的酰胺鍵形成試劑[J].中國醫藥工業雜志,2006,37(12):855-858.
[10]高旭紅,李炳奇.有機合成中的氨基保護及應用(綜述)[J].石河子大學學報:自然科學版,1999,3(1):76-86.
[11]王 閣,曹小輝.Boc或Cbz保護的 L-脯氨醇衍生物的穩定性[J].有機化學,2011,31(6):908-911.
[12]趙 艷,姚金水,戴 罡,等.N-Boc保護基脫除的原理與方法簡介[J].山東輕工業學院學報,2009,23(2):6-7.
[13]朱自紅,曹玉梅.HLPC法測定布洛偽麻那敏片中馬來酸氯苯那敏的含量[J].中國科技博覽,2011(28):602.
[14]殷三福,邱宗蔭.高效液相色譜同時測定布洛偽麻那敏片中布洛芬、鹽酸偽麻黃堿和馬來酸氯苯那敏的含量[J].南方醫科大學學報,2010,30(7):1 704-1 709.
Synthesis of Ibuprofen-Pseudoephedrine Derivatives
Sun Dan1,Lyu Min2,Lyu Zhen2,Li Quan2,Yi Baxian3
(1.Heilongjiang University of Chinese Medicine,Harbin,Heilongjiang,China 150040; 2.Harbin Medisan Pharmaceutical Company,Harbin,Heilongjiang,China 150025; 3.China State Institute of Pharmaceutical Industry,Shanghai,China 201403)
Objective To synthesize and prepare the high-purity derivatives of ibuprofen-pseudoephedrine and demonstrate their structure depending on physicochemical properties and spectral characteristics.M ethods Ibuprofen and pseudoephedrine were used as the starting material,with EDC as condensing agent in esterification and amidation.The finished products were purified by silica gel column chromatography and recrystallization and the structure of the products was detected.Results The purity of each product was more than 99.5%,and their spectral characteristics were consistent with the target compound.Conclusion The synthetic method works easily and the finished products are highly pure.Products can be used as impurity reference for the quality control of compound preparation of ibuprofen-pseudoephedrine.
ibuprofen;pseudoephedrine;esterification;amidation;impurity reference
R914.5;TQ460.3
A
1006-4931(2016)08-0020-04
孫丹(1990-),女,黑龍江哈爾濱人,碩士研究生,研究方向為藥物的質量,(電子信箱)sun1990dan@163.com;李泉(1968-),男,山東淄博人,碩士研究生導師,主要從事新藥研究與開發工作,本文通訊作者,(電話)0451-57355586(電子信箱)hasanlianjinan@aliyun.com。
2015-10-14;
2016-01-15)
