高效液相色譜法分離與測定鹽酸多塞平順反式異構(gòu)體的含量
2016-03-23 02:05:06韓春暉張春泓
中國藥業(yè) 2016年1期
韓春暉,張春泓,欒 爽
(遼寧省大連市藥品檢驗所,遼寧 大連 116021)
高效液相色譜法分離與測定鹽酸多塞平順反式異構(gòu)體的含量
韓春暉,張春泓,欒 爽
(遼寧省大連市藥品檢驗所,遼寧 大連 116021)
目的 建立鹽酸多塞平乳膏中鹽酸多塞平順反式異構(gòu)體分離與含量測定的高效液相色譜法。方法 色譜柱采用Kromasil 100A C18柱(150 mm×4.6 mm,5 μm),流動相為含0.1%三乙胺的0.2 mol/L磷酸二氫鈉溶液-甲醇(65∶35),流速為1.0 mL/min,檢測波長為254 nm,柱溫為50℃。結(jié)果 鹽酸多塞平質(zhì)量濃度在7.90~12.66 μg/mL范圍內(nèi)與峰面積呈良好線性關系(r=0.999 9),平均回收率為98.88%,RSD為1.32%(n=9)。結(jié)論 該方法能良好地分離鹽酸多塞平順反式異構(gòu)體,可作為鹽酸多塞平乳膏中鹽酸多塞平的質(zhì)量控制方法。
高效液相色譜法;鹽酸多塞平;含量測定;異構(gòu)體
鹽酸多塞平乳膏是非類固醇類藥物,主要是利用鹽酸多塞平的H1阻斷作用治療皮炎、濕疹引起的瘙癢,可避免局部使用皮質(zhì)類固醇制劑所帶來的副作用。現(xiàn)行質(zhì)量標準采用紫外分光光度法于296 nm波長處測定含量[1]。本研究中發(fā)現(xiàn),在該波長處有紫外吸收的雜質(zhì),方法專屬性不強,且鹽酸多塞平順反式異構(gòu)體不能良好分離。為此,筆者建立了鹽酸多塞平順反式異構(gòu)體分離的高效液相色譜(HPLC)法,并測定其在鹽酸多塞平乳膏中的含量,現(xiàn)報道如下。
1 儀器與試藥
Waters 2695/2487/2998型高效液相色譜儀;Agilent 1200型高效液相色譜儀;島津LC-20A型高效液相色譜儀;Sartorius MSA225S-100-DA型分析天平(十萬分之一)。鹽酸多塞平對照品(中國食品藥品檢定研究院,批號為100069-201103);鹽酸多塞平乳膏(市售品,批號為2010001,2010002,2008012,規(guī)格為每10 g中含鹽酸多塞平0.5 g);甲醇、磷酸為色譜純,鹽酸、磷酸二氫鈉為分析純,試驗用水為純化水。
2 方法與結(jié)果
2.1 色譜條件與系統(tǒng)適用性試驗
色譜柱:Kromasil 100A C18柱(150 mm×4.6 mm,5 μm);流動相:含0.1%三乙胺的0.2 mol/L磷酸二氫鈉溶液(用 2 mol/L磷酸調(diào) pH至 2.5)-甲醇(65∶35);流速:1.0 mL/min;檢測波長:254 nm;柱溫:50℃;進樣量:20 μL。色譜圖見圖1。理論板數(shù)按順式異構(gòu)體(Z)計算不低于1 500,反式異構(gòu)體(E)與順式異構(gòu)體(Z)峰的分離度大于1.5,乳膏空白基質(zhì)溶液對測定無干擾。
2.2 溶液制備
取鹽酸多塞平對照品適量,精密稱定,置100 mL容量瓶中,加1%鹽酸甲醇溶液溶解并稀釋至刻度,制成每1 mL中含0.1 mg的溶液,得對照品溶液。精密稱取樣品適量(約相當于鹽酸多塞平20 mg),加1%鹽酸甲醇溶液20 mL,置水浴中加熱充分攪拌使鹽酸多塞平溶解,再置冰浴中冷卻1 h以上,使基質(zhì)凝固,取出后迅速濾過,用冷1%鹽酸甲醇溶液洗滌殘渣數(shù)次,用同一濾器濾過,合并濾液與洗液,置100 mL容量瓶中,用1%鹽酸甲醇溶液稀釋至刻度,搖勻,精密量取5 mL置10 mL容量瓶中,加1%鹽酸甲醇溶液稀釋至刻度,搖勻,得供試品溶液。將液態(tài)石蠟、天然脂肪醇、單硬脂酸甘油酯、硬脂酸、平平加A-20、甘油聚乙二醇-75硬脂酸酯、2,6-二叔丁基對甲酚、叔丁基-4-羥基茴香醚、甘油、羥苯乙酯、乙二胺四乙酸二鈉、純化水按處方組分比例稱取適量,加熱至75~80℃,油相與水相等溫混合,攪拌均勻,邊冷卻邊攪拌,直至成霜,作為空白基質(zhì)[2];取約0.4 g空白基質(zhì),按供試品溶液制備方法,即得乳膏空白基質(zhì)溶液。
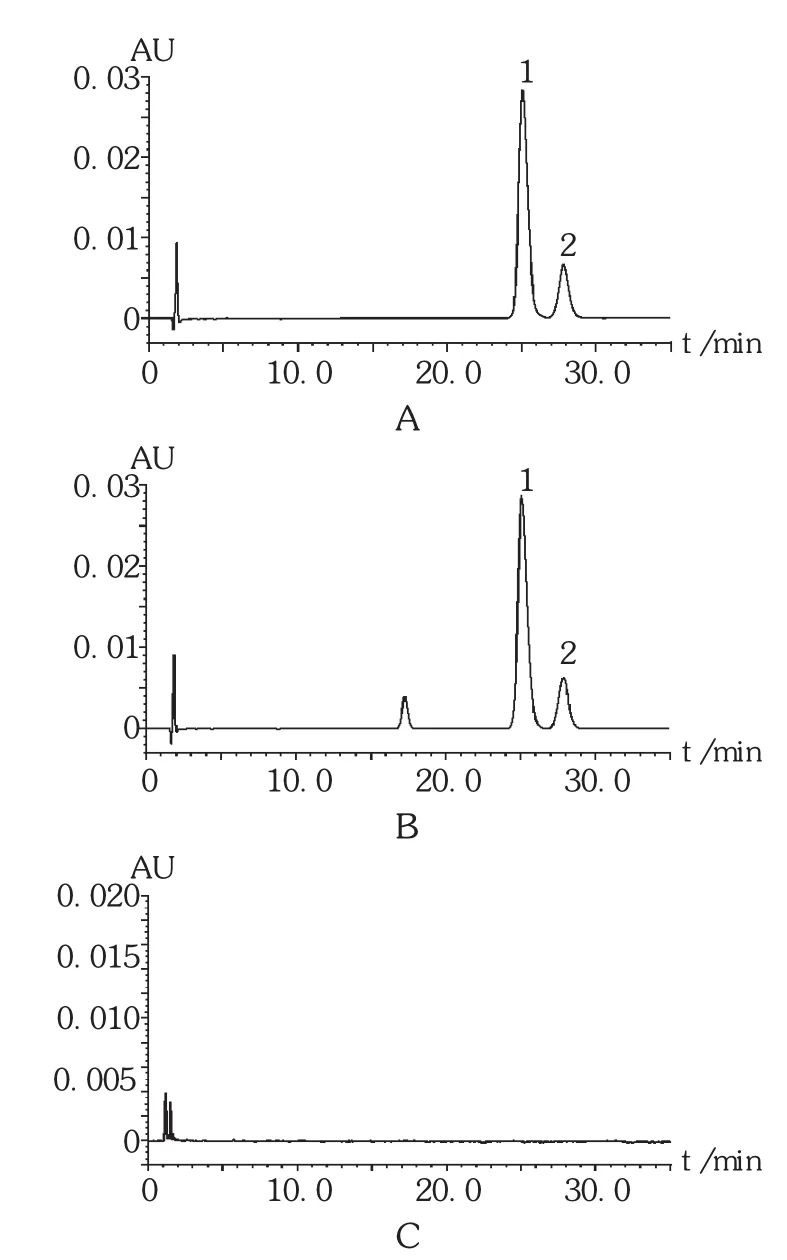
圖1 高效液相色譜圖
2.3 方法學考察
線性關系考察:精密稱取鹽酸多塞平對照品7.90,9.19,10.48,11.45,12.66 mg,分別置 100 mL容量瓶中,用1%鹽酸甲醇溶液溶解并稀釋至刻度,作為系列溶液。分別量取上述溶液各20 μL,注入液相色譜儀,按擬訂色譜條件測定。以質(zhì)量濃度為橫坐標(X)、反式異構(gòu)體(E)與順式異構(gòu)體(Z)峰面積之和(Y)為縱坐標進行線性回歸,得到鹽酸多塞平的線性回歸方程 Y=3.170 6×10-8X-5.803 6×10-5,r=0.999 9(n=5)。結(jié)果表明,鹽酸多塞平質(zhì)量濃度在7.90~12.66 μg/mL范圍內(nèi)與峰面積呈良好線性關系。
精密度試驗:取同一照品溶液,依法連續(xù)進樣6次。結(jié)果的 RSD為1.0%(n=6),表明儀器精密度良好。
穩(wěn)定性試驗:取同一供試品溶液,分別于0,2,4,6,8,12,16,24,48 h時進樣測定。結(jié)果的 RSD為 0.9%(n=9),表明供試品溶液在48 h內(nèi)穩(wěn)定性良好。
重復性試驗:取同一批號的樣品6份,按供試品溶液制備方法制備溶液,依法測定。結(jié)果的 RSD為0.6%(n=6),表明方法重復性良好。
回收率試驗:取鹽酸多塞平原料、輔料,按處方組分比例模擬工藝流程分別制成模擬乳膏,其中已知含量的鹽酸多塞平原料藥分別按標示量的80%,100%,120%加入到模擬乳膏中。依法測定,計算回收率。結(jié)果見表1。
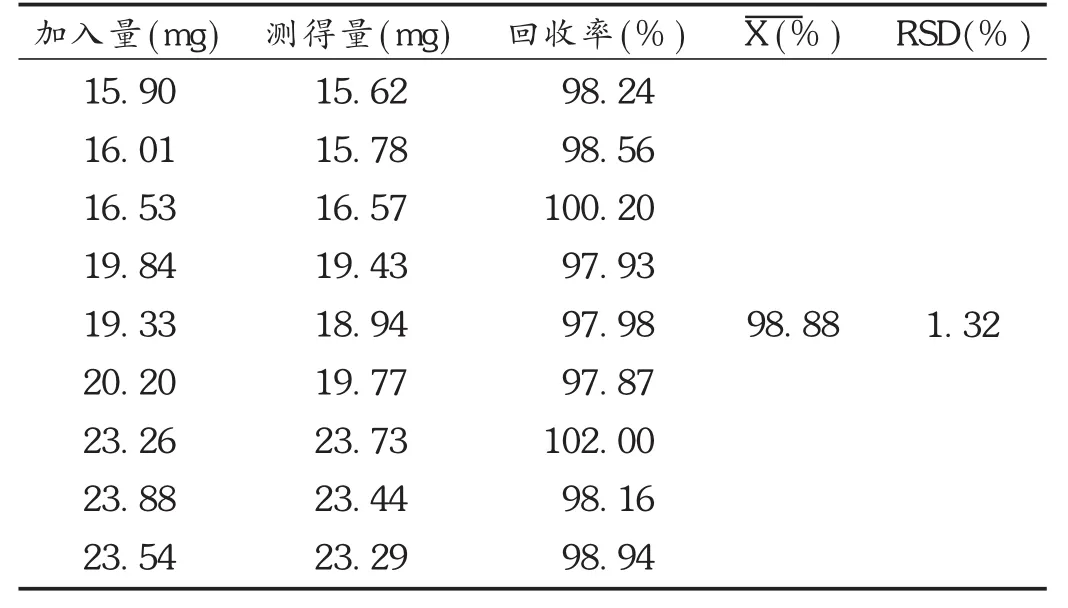
表1 鹽酸多塞平回收試驗結(jié)果(n=9)
2.4 樣品含量測定
取樣品3批(批號為2010001,2010002,2008012),依法制備供試品溶液,按外標法測定并計算含量。結(jié)果3批樣品中含鹽酸多塞平分別為標示量的101.3%,100.5%和100.7%。
3 討論
文獻[3-4]報道,測定鹽酸多塞平含量采用多塞平的紫外最大吸收波長為297 nm。本研究中通過DAD檢測器3D掃描發(fā)現(xiàn),297 nm波長處峰純度不高。通過分析國內(nèi)企業(yè)的生產(chǎn)工藝得知,鹽酸多塞平的主要工藝雜質(zhì)為羥基物,該物質(zhì)于297 nm波長處有較強紫外吸收,嚴重干擾鹽酸多塞平的測定。故選擇測定波長為鹽酸多塞平吸收較強且雜質(zhì)吸收較弱的254 nm。
鹽酸多塞平原料藥為反式異構(gòu)體(E)與順式異構(gòu)體(Z)的混合物。早期含量測定中未將順反式異構(gòu)體分離[5],導致色譜峰出現(xiàn)明顯肩峰。有研究報道,使用HPLC法[6]和毛細管氣相色譜法[7]測定順式異構(gòu)體含量。本研究中通過優(yōu)化色譜條件,將順反式異構(gòu)體完全分離,排除了測定過程中的相互干擾,相關研究尚未見有報道。
流動相的水相中加入0.1%的三乙胺,有效地改善了峰拖尾現(xiàn)象。試驗表明,在主成分與各雜質(zhì)的分離中順、反式異構(gòu)體峰最難分離,故使用順、反式異構(gòu)體峰的分離度進行系統(tǒng)適用性試驗。當順、反式異構(gòu)體峰的分離度大于1.5時,其他雜質(zhì)均可獲得良好分離。
本研究中采用了另外3個品牌的色譜柱,如AMERITECH Accurasil C18柱(150 mm×4.6 mm,5 μm),Dilkma Platisil C18柱(150 mm×4.6 mm,5 μm),ThermoHyparcil GOLD C18柱(250 mm×4.6 mm,5 μm),分別在Waters 2695/2996型、Agilent 1200型與島津 LC-20A型液相色譜儀上進行方法耐用性試驗考察。結(jié)果提示,供試品溶液色譜圖中順反式異構(gòu)體之間及與相鄰雜質(zhì)峰間分離度均大于1.5,耐用性良好。
[1]國家藥典委員會.國家食品藥品監(jiān)督管理局《國家藥品標準》新藥轉(zhuǎn)正標準第 34冊[M].北京:人民衛(wèi)生出版社,2003:193.
[2]唐 芳,丁登峰,周金玉.復方鹽酸多塞平乳膏的制備與質(zhì)量控制[J].中國藥房,2004,15(1):26-28.
[3]周利娟,艾嗚哲,谷亦平.高效液相色譜法測定鹽酸多塞平乳膏中鹽酸多塞平含量[J].醫(yī)藥導報,2007,26(8):938.
[4]宋興發(fā),高 山.高效液相色譜法測定鹽酸多塞平片含量[J].醫(yī)藥導報,2009,28(10):1 359-1 360.
[5]張晨華.反相高效液相色譜法測定鹽酸多塞平片的含量[J].中國藥師,2007,10(12):1 220-1 221.
[6]郝榮華.高效液相色譜法測定鹽酸多塞平順式異構(gòu)體限度[J].藥學與臨床研究,2008,16(3):242-244.
[7]周利娟,艾鳴哲,谷亦平.毛細管氣相色譜法測定鹽酸多塞平順式異構(gòu)體含量[J].醫(yī)藥導報,2006,25(4):344-345.
Separation and Determination of ZE-Doxepin Hydrochloride by HPLC
Han ChunHui,Zhang ChunHong,Luan Shuang
(Dalian Institute for Drug Control,Dalian,Liaoning,China 116021)
Objective To establish a method for the separation and determination of Doxepin Hydrochloride in Doxepin Hydrochloride Cream by HPLC.Methods Chromatography was performed on a Kromasil 100A C18(150 mm×4.6 mm,5 μm)column.The mobile phase was consisted of 0.1% triethylamine in 0.2 mol/L sodium dihydrogen phosohate solution-methanol(65∶35).The flow rate was 1.0 mL/min.The detection wavelength was 254 nm.The column temperature was 50℃.Results The linear response range was 7.90~12.66 μg/mL for Doxepin Hydrochloride(r=0.999 9).The recovery of Doxepin Hydrochloride was 98.88%(RSD=1.32%,n=9).Conclusion The method can separate ZE-doxepin hydrochloride,which was suitable for the determination of Doxepin Hydrochloride in doxepin hydrochloride cream.
HPLC;doxepin hydrochloride;determination;isomer
R927.11;R971+.43
A
1006-4931(2016)01-0061-03
韓春暉,男,碩士研究生,主管藥師,主要從事藥物分析及藥品質(zhì)量標準研究,(電話)0411-84255311(電子信箱)luanshuangdl@163.com。
2015-07-28)
