產肌酸水解酶基因工程菌的構建*
2015-12-16 02:27:27楊波王滾蔣芬劉冠蘭楊成段紹斌蔣云生
中國現代醫學雜志 2015年29期
楊波,王滾,蔣芬,劉冠蘭,楊成,段紹斌,蔣云生
(1.南華大學附屬第一醫院腎內科,湖南 衡陽 421001;2.長沙醫學院臨床醫學系,湖南 長沙 410219;3.中南大學湘雅二醫院腎內科,湖南 長沙 410011)
·論著·
產肌酸水解酶基因工程菌的構建*
楊波1,王滾1,蔣芬1,劉冠蘭1,楊成2,段紹斌3,蔣云生3
(1.南華大學附屬第一醫院腎內科,湖南 衡陽 421001;2.長沙醫學院臨床醫學系,湖南 長沙 410219;3.中南大學湘雅二醫院腎內科,湖南 長沙 410011)
目的構建肌酸水解酶的重組質粒并轉化至大腸桿菌,研究其蛋白的表達及活性。方法①根據Genbank數據庫已知的肌酸水解酶(黃桿菌U-188)的基因序列,由上海吉凱生物有限公司進行生物合成,得到肌酸水解酶基因片段b;②根據肌酸酶的基因序列設計引物,聚合酶鏈反應(PCR)擴增,進行雙酶切的回收和純化,與質粒GV296酶切后進行連接,構建重組質粒GV296-b,分別轉化至大腸桿菌DH5a,篩選測序鑒定;③提取重組質粒GV296-b,轉入至大腸桿菌BL21(DE3),進行菌液PCR鑒定;④工程菌GV296-b/BL21(DE3),向菌液中加入異丙基硫代-β-D-半乳糖苷(IPTG)誘導其蛋白表達,并制備粗酶液進行十二烷基硫酸鈉-聚丙烯酰胺(SDS-PAGE)分析;⑤將工程菌GV296-b/BL21(DE3)于含特定濃度肌酸的培養基中培養24 h,取培養液測定肌酸濃度。結果①成功構建重組質粒GV296-b,將上述重組質粒進行雙酶切。電泳顯示,目的片段大小約為0.783和1.212 kb,與理論值相符;②重組質粒GV296-b,轉化至E.coliBL21(DE3)感受態細胞中,經IPTG誘導后實現高效表達。工程菌GV296-b/BL21(DE3)表達肌酸酶,具有肌酸酶活性,按基因序列圖分析肌酸水解酶403個氨基酸,經SDS-PAGE分析酶蛋白的分子量約為43 kD。結論成功構建的工程菌GV296-b/ BL21(DE3)能高效誘導肌酸酶的表達,具有肌酸酶活性。
肌酸水解酶;基因工程菌;肌酸;肌氨酸
慢性腎功能衰竭(簡稱慢性腎衰)幾乎是所有泌尿系疾病的共同歸宿,也可繼發于其他全身性疾病,如原發性高血壓和糖尿病,后者約有43%并發慢性腎衰。據國際腎臟病協會統計,慢性腎衰的人群發病率約為100~200/100萬,美國成年人中慢性腎衰的發病率高達7.6%,而日本的發病率更高達996/100萬[1-2]。我國慢性腎臟病的發病率約為10.9%[3],隨著人口老齡化速率逐年加快,慢性腎衰的發病率也隨之增加,嚴重威脅著人們的健康及生命。對于慢性腎衰終末期的治療,現主要依靠血液透析、腹膜透析和腎臟移植進行腎臟替代治療。目前,對腎臟替代前期治療采用非透析治療,其經典方法是胃腸道吸附劑,如氧化淀粉及活性碳制劑。消化道酶療法的基本方法是采用微膠囊包裹尿素酶及吸附劑,使肌酐、尿素等及其他毒素物質進入微膠囊內得到吸附分解。同時興起的是腸道細菌療法,最初選用自然菌種,如腸道菌和低毒土壤菌,制成活菌制劑攝入消化道,分泌分解酶降解腸道內尿毒素。但上述自然菌存在產酶特性不穩定及菌的安全性問題,以致人們致力于基因工程菌的研究。肌酐在肌酐酶催化下生成肌酸,后者在肌酸酶催化下,降解為肌氨酸。本實驗將肌酸酶的基因轉入特定細菌中,再將這含有某種酶的特定菌種制成生物制劑,攝入腎衰患者的消化道,將患者腸道中的肌酸(由肌酐酶催化肌酐生成)降解為肌氨酸。肌氨酸不但無毒,而且為營養物質,從而達到治療的目的。
1 材料與方法
1.1 材料
大腸桿菌DH5a(E.coliDH5a)、E.coliBL21(DE3)為南華大學微生物實驗室保存,質粒PET28a(+)(GV296),構于上海吉凱生物有限公司,DNA限制性內切酶為美國Biolabs公司生產,T4 DNA Ligase為美國Thermo公司生產,2×Taq Master Mix為北京康為世紀生物科技有限公司生產,高純度質粒小提試劑盒及DNA凝膠回收試劑盒為北京天根生化科技有限公司生產。
1.2 實驗方法
1.2.1 肌酸酶目的基因片段的構建根據Genbank數據庫已知的肌酸酶(黃桿菌U-188)D14464.1的基因序列,由上海吉凱生物有限公司進行生物合成,得到肌酸酶目的基因模板b。
1.2.2 聚合酶鏈反應(polymease chain reaction,PCR)擴增根據黃桿菌肌酸水解酶的基因序列設計引物,在正向引物中添加酶切位點BamHⅠ,在反向引物中添加酶切位點XhoⅠ,以便以后的克隆和表達。引物設計為正向引物:AGCAAATGGGTCGC GGATCCATGCAAATGCCCAAAACAC;反向引物:T GGTGGTGGTGGTGCTCGAGTTATTTGCGGATGATG TTTC。反應體系(25μl):模板b 1μl,正向引物0.5μl,反向引物0.5μl,2×Taq Master Mix 12.5μl,ddH2O 10.5μl,總體積為25μl。PCR反應條件為:94℃預變性5 min,94℃變性30 s,55℃退火30 s,72℃延伸80s,共30個循環,72℃繼續延伸7 min。反應結束后4℃保存。
1.2.3 PCR產物及質粒的酶切用BamHⅠ和XhoⅠ對PCR產物和GV296進行雙酶切。酶切體系(50μl):DNA 10μl,BamHⅠ1μl,XhoⅠ1μl,Cursmart Buffer 5μl,ddH2O 33μl。37℃反應3 h,65℃滅活10min。
1.2.4 酶切產物電泳并割膠回收肌酸酶目的基因片段的PCR產物及質粒GV296雙酶切后,用DNA凝膠回收試劑盒回收DNA片段。
1.2.5 肌酸酶基因片段b與質粒GV296的連接回收純化的肌酸酶DNA片段b及質粒GV296酶切回收片段,按照1∶5加樣進行連接反應。T4連接酶反應體系(20μl):T4連接酶1μl,10×連接Buffer 2μl,回收純化的肌酸酶DNA片段5μl,質粒GV296酶切回收產物1μl,ddH2O 11μl。22℃反應10 min,65℃滅活10 min。重組質粒GV296-b轉化E.coli感受態細胞。
1.2.6 轉化子挑選與鑒定重組質粒GV296-b轉化子使用菌落PCR、雙酶切及DNA測序篩選陽性克隆菌。先用菌落PCR初步篩選陽性克隆,然后抽提重組質粒,進行雙酶切和DNA測序確定。①菌落PCR:在無菌條件下用接種環在平板上挑取單菌落(選取直徑>1 mm的菌落),分別挑取1個單菌落于1 ml卡那霉素抗性LB培養基離心管中培養,共培養8管,37℃、220 r/min振蕩2 h。以通用引物T7 Promoter Primer作為PCR及測序引物。T7引物序列的正向引物:GCTGGAGCGTACCCTGTTC;反向引物:TGCTAGTTATTGCTCAGCGG。反應體系(12.5μl):菌液1.5μl,T7正向引物0.25μl,T7反向引物0.25 μl,2×Taq Master Mix 6.25μl,ddH2O 4.25μl,總體積12.5μl。反應條件為:94℃預變性10 min,94℃變性30 s,55℃退火30 s,72℃延伸80 s,共30個循環,72℃繼續延伸7 min。PCR反應結束后,置入4℃冰箱保存。將PCR產物進行1.2%瓊脂糖凝膠電泳,初步篩選陽性克隆菌。②重組質粒GV296-b雙酶切及測序:取10μl菌落PCR篩選到的陽性克隆菌液接種于4 ml卡那霉素抗性LB培養基中培養,37℃、220 r/min振蕩培養過夜(約12 h)。用質粒提取盒抽提質粒,進行雙酶切。將菌落PCR及酶切均顯示為陽性的重組質粒GV296-b轉化子菌液,送至上海生工生物有限公司測序。
1.2.7 重組質粒GV296-b的鑒定及誘導表達測序正確的陽性轉化子GV296-b,提取質粒后,轉入至大腸桿菌BL21(DE3),進行轉化及菌液PCR,構建表達工程菌GV296-b/BL21(DE3)。根據測序比對正確的肌酸酶序列設計引物,在引物中分別添加酶切位點HindⅢ和XbaⅠ。引物序列為:正向引物CCCAAGCTT-CAGCTGTTGCGCCAGAC;反向引物:GCTCTAGA-GTGGCACGGAGGTTCG。PCR反應結束后,產物進行1.2%瓊脂糖凝膠電泳,篩選陽性克隆菌。采用異丙基硫代-β-D-半乳糖苷(isopropylbeta-D-thiogalactopyranoside,IPTG)誘導PCR鑒定陽性的表達工程菌GV296-b/BL21(DE3)蛋白的表達。細菌超聲波粉碎后,采用非連續變性電泳,12%濃度的分離膠,5%濃度的濃縮膠。
1.2.8 表達工程菌分解肌酸的體外實驗從平板上挑取重組菌GV296-b/BL21(DE3)的單菌落接種于2 ml卡那霉素抗性LB培養基中,37℃、220 r/min揺菌培養過夜。次日按1∶100轉種于5 ml卡那霉素抗性LB培養基中,30℃、220r/min培養2h。無菌條件下加入IPTG至終濃度1 mmol,繼續培養4 h,收集菌體,先用生理鹽水洗滌,再用生理鹽水重懸菌體,并用麥氏比濁法調整懸菌液約為1.5×108cfu/ml。取200μl懸菌液加入800μl含特定濃度肌酸的LB培養基中培養,取200μl大腸桿菌DH5a(菌濃度同上)懸菌液,加入800μl含特定濃度肌酸的LB培養基,作為對照組。37℃、220 r/min培養24 h,13 000 r/min離心2 min,取上清液用生化分析儀檢測肌酸濃度。取200μl生理鹽水加入上述培養基中作為對照組,培養24 h后,同樣離心取上清液測肌酸濃度。每組設5個樣本。
1.3 統計學方法
采用SPSS 20.0統計軟件進行數據分析,計量資料以均數±標準差(±s)表示,用單因素方差分析(one-way,ANOVA),多重比較用最小顯著法(LSD),P<0.05為差異有統計學意義。
2 結果
2.1 PCR擴增的肌酸酶目的基因
根據Genbank數據庫已知的肌酸水解酶(黃桿菌U-188)基因序列,由上海吉凱公司進行生物合成模板b,設計引物,進行PCR擴增,可得到大小約1 252bp的片段,與預期大小一致。見圖1。

圖1 肌酸酶基因PCR產物電泳圖
2.2 PCR鑒定重組質粒GV296-b
重組質粒GV296-b轉化至大腸桿菌DH5a感受態細胞,挑取轉化子1~8號,進行菌液PCR,產物經1.2%瓊脂糖凝膠電泳。泳道5~12為1~8號轉化子,經PCR鑒定得到大小約529 bp的片段,與肌酸酶的基因大小相符(見圖2)。將PCR鑒定陽性的產物送測序公司測序,證實肌酸酶的基因片段與Genbank報道的基因序列一致(見圖3)。

圖2 重組質粒GV296-b的菌落PCR測定
2.3 表達工程菌GV296-b/BL21(DE3)的十二烷基硫酸鈉-聚丙烯酰胺分析
表達工程菌GV296-b/BL21(DE3)經IPTG誘導表達,終濃度為1.0 mmol,30℃、200 r/min離心4 h后,4℃、13 000 r/min離心1 min,收集沉淀。用磷酸鹽緩沖溶液(phosphate buffer saline,PBS)洗滌菌體,超聲碎菌體,離心收集上清液和沉淀。分別加入4×十二烷基硫酸鈉(sodium dodecyl sulfate,SDS)上樣緩沖液,100℃、煮沸10 min,用于十二烷基硫酸鈉-聚丙烯酰胺(sodium dodecyl sulfate-polyacrylamide gelelectrophoresis,SDS-PAGE)上樣,凝膠成像照相。泳道2、4分別為誘導的1、2管沉淀;泳道3、5分別為誘導的1、2管上清液;泳道6、8為未誘導的1、2管沉淀;泳道7、9為未誘導的1、2管上清液。分析可見泳道3、5有目的蛋白表達,分子量約為43 kD,與預期相符合(見圖4)。
2.4 表達工程菌分解肌酸活性的測定
表達工程菌GV296-b/BL21(DE3)經IPTG誘導后,取菌液與特定濃度的肌酸作用24 h。作用后表達工程菌組上清液肌酸濃度為(403.4±16.4)μmol/L,與對照組比較[(555.6±11.9)μmol/L]顯著下降,差異有統計學意義(P<0.01);DH5a組為(548.7± 13.5)μmol/L,與陰對照組比較,差異無統計學意義(P>0.05)。

圖3 肌酸酶基因片段的測序圖
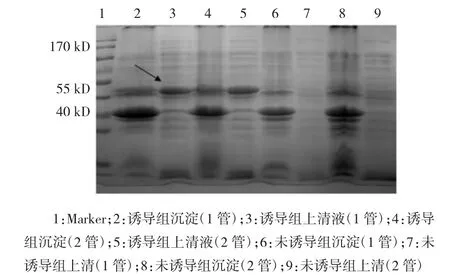
圖4 重組肌酸酶誘導表達的SDS-PAGE分析
3 討論
在慢性腎衰狀態下,機體發生氧化應激反應也相對增加,活性氧和氫氧基及其他自由基生成增多,肌酐與其作用生成氫氧化肌酐,后者在各種酶的作用下最后生成甲基胍[4]。甲基胍升高是引起惡心、嘔吐、腹瀉、貧血、糖耐量降低、血漿纖維蛋白增高及裂解活性下降、鈣吸收減少、胃十二指腸潰瘍和出血、抽搐和意識障礙等的主要原因,還可引起胰腺等分泌減少、紅細胞自溶,并且對淋巴細胞的DNA合成有抑制作用,同時抑制去甲腎上腺素在交感神經突觸小泡中運輸。以前有學者將肌酐水解酶克隆入腸道益生菌,使肌酐在腸道內降解成肌酸[5]。然而肌酐經肌酐水解酶生成的肌酸,可通過肝腸循環,經腸道進一步重吸收進入血液,在體內再次代謝轉化成肌酐,并未解決根本問題。肌酐在肌酐水解酶的作用下生成肌酸,肌酸在肌酸水解酶作用下進一步生成肌氨酸和尿素。本實驗室早期成功構建了肌酐水解酶基因重組工程菌,此次構建含肌酸水解酶的重組菌,表達肌酸水解酶,旨在將肌酐降解肌酸后,進一步降解為肌氨酸。
本實驗中,根據Genbank黃桿菌U-188的基因序列進行生物合成肌酸酶目的基因模板,PCR擴增分別得到大小約1.252 kb的肌酸酶目的基因片段。將上述擴增目的片段與質粒GV296連接后,經測序鑒定擴增片段的基因與已知Genbank上的基因完全一致。通過測序比對得到肌酸酶的基因片段大小約為1.212 kb,經酶切后同樣證實為肌酸酶的基因片段,顯示該基因克隆成功。實驗中使用的質粒PET-28a(+)是帶有T7 lac啟動子的載體,其T7啟動子下游有一段lac阻遏蛋白(lacI)操縱子序列編碼表達lacI。lacI可以作用于宿主染色體上T7RNA聚合酶前的lacUV5啟動子并抑制其表達,也作用于載體T7 lac啟動子,以阻斷任何T7RNA聚合酶導致的目的基因轉錄,IPTG通過與lacI結合而保持lac操縱子轉錄開放。實驗結果顯示,工程菌有肌酸酶蛋白的表達,并且具有分解肌酸活性。
腸道細菌療法中極其重要地是選擇高效低毒菌,因為外來菌攝入消化道后,可能引起宿主腸道微生態改變,出現新的菌群失調;同時其還可能攜帶某些耐抗菌素的質粒,這些質粒可能會轉入到其他腸道菌種中,導致耐抗生素細菌的感染,對原本免疫力低下的尿毒癥患者產生嚴重的感染性疾病。大腸桿菌是人和動物腸道中的正常棲居菌,其雖然為條件致病菌,但大部分血清型大腸桿菌為人體正常菌群,無異位的大腸桿菌沒有致病性。PRAKASH等[6]研究表明,微囊化的基因修飾大腸桿菌DH5a對動物(老鼠)無致病性。大腸桿菌與代謝有關的酶種類很多,這些酶的產生與外環境息息相關。對于尿毒癥患者來說,許多代謝廢物不能充分經腎臟排泄而蓄積于體內,大量的代謝廢物通過豐富的腸壁血管進入腸道,使腸腔內呈高代謝廢物水平狀態,為細菌的生長繁殖提供豐富的營養及充足的條件從而誘導細菌生長;同時這種環境也能誘導大腸桿菌產生多種酶降解肌酐、尿素氮和尿酸。大腸桿菌表達系統是目前最常用的外源蛋白表達系統,其不僅具有遺傳背景清楚、操作簡便、容易培養,還具有能快速大規模地生產目的蛋白的優點[7]。其表達外源基因產物的水平遠遠高于其他基因表達系統,表達的目的蛋白甚至超過細菌總蛋白量的30%,使之成為人們克隆表達外源基因的主要菌株。
至今為止,基因工程菌宿主菌主要為大腸桿菌,然而大腸桿菌為條件致病菌,存在一個潛在性的問題。大腸桿菌是活菌,攝入消化道后在腸道繁殖,擾亂腸道微生態,使腸道菌群比例改變,造成細菌及毒素移位,導致慢性腎衰微炎癥改變[8]。因此考慮下一步采用益生菌做為基因工程菌的宿主菌,有望為慢性腎衰腸道細菌的治療解決安全性問題。近20年來,人們逐漸致力于乳酸桿菌的生物學性狀和基因組學的研究,并且通過基因工程的方法,對其進行遺傳修飾,開發構建一系列乳酸桿菌基因表達系統,將其作為基因工程菌用于疫苗研制。本實驗成功構建肌酸水解酶的重組質粒菌,為下一階段構建含有肌酸水解酶的乳酸桿菌基因工程菌奠定基礎。作為毒素的肌酐,在轉化為肌酸后,進一步轉化為營養物質(肌氨酸),為延緩腎功能衰竭及針對慢性腎衰的治療提供新的思路及治療方法。
[1]CHEN W,CHEN W,WANG H,et al.Prevalence and risk factors associated with chronic kidney disease in an adult population from southern China[J].Nephrology Dialysis Transplantation, 2009,24(4):1205-1212.
[2]陳孝文,梁東.慢性腎衰竭[M].北京:中國醫藥科技出版社,2006: 486-488.
[3]ZHANG L,WANG F,WANG L,et al.Prevalence of chronic kidney disease in China:a cross-sectional survey[J].The Lancet, 2012,379(9818):815-822.
[4]車文體,董力,鄒貴勉.結腸透析聯合藥用活性炭對慢性腎功能衰竭患者鈣磷和尿酸的吸附作用[J].國際移植與血液凈化雜志,2012, 10(6):23-25.
[5]程新,張彥新,周桂鳳,等.產肌酐水解酶基因工程菌的構建[J].生物醫學工程研究,2009,28(4):299-302.
[6]PRAKASH S,CHANG TM.Growth and survival of renal failure rats that received oral microencapsulated genetically engineered E.coliDH5 cells for urea removal[J].Artif Cells Blood Substit Immobil Biotechnol,1998,26(1):35-51
[7]NUC P,NUC K.Recombinant protein production in escherichia eoli[J].Postepy Biochem,2006,52(4):448-456.
[8]WANG F,ZHANG P,JIANG H,et al.Gut bacterial translocation contributes to microinflammation in experimental uremia[J]. Dig Dis Sci,2012,57(11):2856-2862.
(申海菊 編輯)
Construction of creatinase gene engineering bacteria*
Bo YANG1,Gun WANG1,Fen JIANG1,Guang-lan LIU1,Cheng YANG2, Shao-bin DUANG3,Yun-sheng JIANG3
(1.Department of Nephrology,the First Affiliated Hospital,University of South China, Hengyang,Hunan 421001,P.R.China;2.Clinical Department,Changsha Medical College, Changsha,Hunan 410219,P.R.China;3.Department of Nephrology,the Second Xiangya Hospital,Central South University,Changsha,Hunan 410011,P.R.China)
【Objective】To construct a plasmid containing creatinase gene,transform it intoEscherichia coliand study its expression and enzyme activity.【Methods】The creatinase(Flavobacterium sp.U-188)gene sequence was biosynthesized according to the GenBank database to get the gene fragment of creatinase b.The primers of creatinase were designed according to the gene sequence.The gene was amplified by PCR.Then the DNA was extracted after double enzyme digestion and purified.The purified DNA was cloned to GV296 to construct the recombinant plasmid GV296-b,which were transformed intoE.coliDH5a for screening and identification.The recombinant plasmid GV296-b was extracted and transformed toE.coliBL21(DE3)for PCR identification of bacterial liquid.The inducer isopropyl-beta-D-thiogalactopyranoside(IPTG)was added into the engineering strain GV296-b/BL21(DE3)to induce protein expression,and SDS-PAGE was conducted. The GV296-b/BL21(DE3)was cultured in the culture medium at a particular concentration of creatinine acid for 24 hours,and then the concentration of creatinine acid in the culture fluid was determined.【Results】①The recombinant plasmid GV296-b was successfully constructed and double digested.Electrophoresis showedthe two restriction enzyme fragments were 0.783 kb and 1.212 kb which were consistent with the theoretical values.②The reconstructed plasmid GV296-b was transformed intoE.coliBL21(DE3)competent cells.The bacterium was induced by IPTG and achieved efficient expression.The bacterium GV296-b/BL21(DE3)expressed active creatinase which was composed of 403 AA in gene sequence mapping with the molecular weight of approximate 43 kD by SDS-PAGE.【Conclusions】The engineering strain GV296-b/BL21(DE3)has been successfully constructed,and the expression of the active creatinase has been efficiently induced.
creatinase;gene engineering bacteria;creatine;sarcosine
Q814
A
1005-8982(2015)29-0012-06
2015-06-16
湖南省自然科學基金(No:13JJ3082)
