朗格漢斯細(xì)胞組織細(xì)胞增生癥6例臨床病理分析
2015-10-26 06:19:40邱立柳玉紅溫壽青汪春福陳蕾龔靜青
中國(guó)醫(yī)藥導(dǎo)報(bào) 2015年14期
邱立 柳玉紅 溫壽青 汪春福 陳蕾 龔靜青
廣東省深圳市寶安區(qū)人民醫(yī)院,廣東深圳518101
朗格漢斯細(xì)胞組織細(xì)胞增生癥6例臨床病理分析
邱立柳玉紅溫壽青汪春福陳蕾龔靜青
廣東省深圳市寶安區(qū)人民醫(yī)院,廣東深圳518101
目的探討朗格漢斯細(xì)胞組織細(xì)胞增生癥(LCH)臨床病理特點(diǎn)、免疫表型及預(yù)后特點(diǎn)。方法分析6例兒童LCH的臨床資料及組織病理學(xué)特點(diǎn),同時(shí)用免疫組化染色觀察LCH的免疫表型,并結(jié)合文獻(xiàn)復(fù)習(xí)。結(jié)果男性4例,女性2例,年齡最小23 d,最大13歲。組織學(xué)改變:受累器官內(nèi)朗格漢斯細(xì)胞彌漫增生,細(xì)胞中等偏大,核呈卵圓形、腎形,可見核折疊。免疫組化:朗格漢斯細(xì)胞S-100、CD68和CD1α(+),LCA、CK、CD3、CD5、CD20(-)。結(jié)論LCH臨床表現(xiàn)復(fù)雜,診斷需要結(jié)合臨床表現(xiàn)、影像學(xué)檢查及組織病理學(xué)檢查。該病患兒預(yù)后差別較大,取決于發(fā)病年齡、受累器官多少、器官功能受損嚴(yán)重性。
朗格漢斯組織細(xì)胞增生癥;病理分析;免疫表型
朗格漢斯細(xì)胞組織細(xì)胞增生癥(Langerhans cell histiocytosis,LCH)是一種因朗格漢斯細(xì)胞(Langerhans cell)克隆性彌漫增生浸潤(rùn),導(dǎo)致器官功能障礙為特征的罕見腫瘤性疾病,曾用名嗜酸性肉芽腫、組織細(xì)胞增生癥X、Langerhans細(xì)胞肉芽腫病等,其發(fā)病率約為5/100萬,在任何年齡階段均可發(fā)病,但主要患者人群見于低齡兒童,男性較為多見(男女之比約為1.6~2∶1)。因其起病癥狀較隱匿,臨床表現(xiàn)復(fù)雜多樣,病情多變,容易導(dǎo)致臨床醫(yī)生的誤診。本文收集廣東省深圳市寶安區(qū)人民醫(yī)院(以下簡(jiǎn)稱“我院”)診治的6例LCH,對(duì)其臨床體征、病理學(xué)特征以及免疫組化表達(dá)情況進(jìn)行整理分析,結(jié)合文獻(xiàn)探討其發(fā)病機(jī)制、病理診斷、鑒別診斷及預(yù)后,以提高對(duì)該病的認(rèn)識(shí)。
1 資料與方法
1.1一般資料
收集6例LCH患兒均為2006年2月~2014年2月收治入我院的病理確診病例,查閱并整理臨床病歷資料并電話隨訪患兒情況。
1.2方法
所有標(biāo)本經(jīng)4%中性甲醛溶液固定12 h,骨組織預(yù)先經(jīng)脫鈣處理,常規(guī)脫水、石蠟包埋切片,行HE染色,光鏡觀察。采用羅氏(Roche)全自動(dòng)免疫組化儀。一抗包括CD68、S-100、CD1α、LCA、CK、CD20、CD79a、CD3、ALK和Ki-67,均為單克隆即用型抗體,以上試劑均購(gòu)自廣州安必平醫(yī)藥科技有限公司。PBS液替代一抗做陰性對(duì)照,用標(biāo)準(zhǔn)陽(yáng)性片做陽(yáng)性對(duì)照,染色步驟過程按試劑盒及機(jī)器標(biāo)準(zhǔn)程序完成。細(xì)胞質(zhì)和/或細(xì)胞核有深棕色細(xì)顆粒附著視為陽(yáng)性表達(dá)。
2 結(jié)果
2.1臨床資料
6例患兒,男4例,女2例,發(fā)病年齡為23 d~13歲。6例均為首次發(fā)病,其中4例為單發(fā)病灶,2例為多發(fā)病灶。見表1。

表16 例朗格漢斯細(xì)胞組織細(xì)胞增生癥患者臨床資料
2.2組織學(xué)觀察
光鏡下可見朗格漢斯細(xì)胞彌漫性增生,分布疏密不均;細(xì)胞呈圓形、卵圓形,中等大小,胞漿偏少,胞質(zhì)粉染,界限不清;核呈圓形、卵圓形、腎形或不規(guī)則形,部分可見核溝、核凹陷及核折疊,核膜薄,染色質(zhì)細(xì)膩,核仁不明顯。病例2可見散在多核巨細(xì)胞分布其間(圖1A);病例5腫瘤細(xì)胞核可見明顯的核溝和不規(guī)則扭曲,異型性不明顯,核分裂象少見(圖1B),病變區(qū)域內(nèi)可見較多嗜酸性粒細(xì)胞、淋巴細(xì)胞分布其間,間質(zhì)富于小血管。
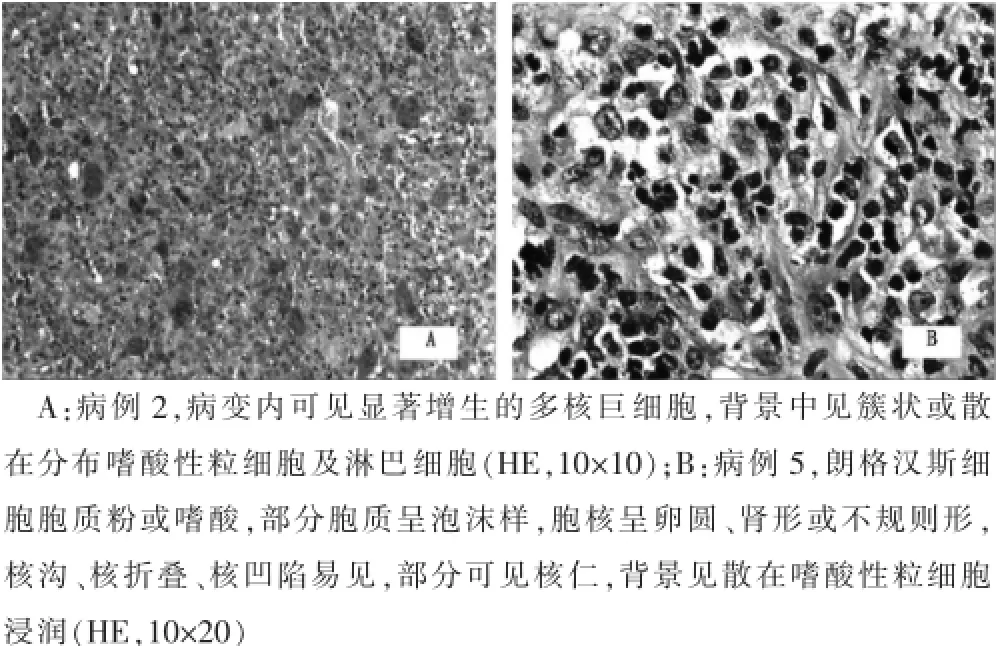
圖1 朗格漢斯細(xì)胞組織細(xì)胞增生癥光鏡下組織學(xué)圖像(HE染色)
2.3免疫組化
6例病變內(nèi)朗格漢斯細(xì)胞CD1α(圖2A)、CD68和S-100(圖2B)均為(+);病例1、5 p53呈弱(+),病例2、3、4、6為(-);各例Ki-67增殖指數(shù)為5%~10%;各例LCA、CK、CD20、CD79a、CD3、ALK標(biāo)記均為(-)。
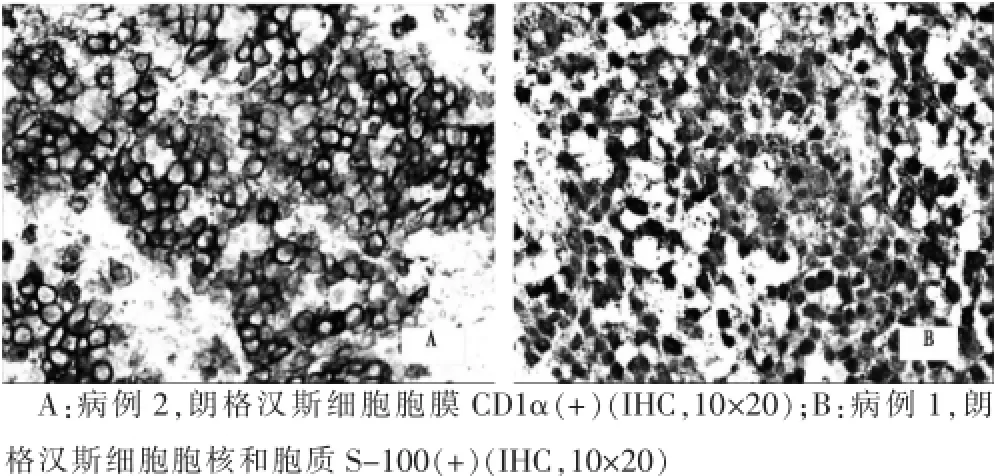
圖2 朗格漢斯細(xì)胞組織細(xì)胞增生癥光鏡下免疫組化圖像
2.4病例隨訪情況
本組6例患兒中2例(病例3、病例6)在6個(gè)月內(nèi)死亡(年齡均<1歲,病例6為多發(fā)病灶),1人(病例4)放棄治療出院,電話追訪得知患兒在1年內(nèi)死亡。其余3例經(jīng)綜合治療后目前暫未復(fù)發(fā),患者一般情況尚可,還需繼續(xù)觀察。
3 討論
朗格漢斯細(xì)胞是一類正常狀態(tài)下存在于皮膚和黏膜基底層的細(xì)胞,屬于組織細(xì)胞系統(tǒng),是一種特化的樹突狀細(xì)胞[1]。有觀點(diǎn)認(rèn)為,其處在樹突狀細(xì)胞發(fā)育過程中的不成熟階段,一旦激活,可將特異性抗原呈遞給C細(xì)胞[2]。LCH原來曾稱為“組織細(xì)胞增生癥X”和“Langerhans細(xì)胞肉芽腫病”,是一類起源于朗格漢斯細(xì)胞并繼而克隆性增生導(dǎo)致的腫瘤性疾病,其腫瘤細(xì)胞保持了朗格漢斯細(xì)胞的免疫表型和超微結(jié)構(gòu)特征。在2008年WHO腫瘤分類中,LCH被歸于組織細(xì)胞和樹突狀細(xì)胞腫瘤中的一類。
3.1發(fā)病機(jī)制
研究發(fā)現(xiàn),LCH中朗格漢斯細(xì)胞呈單克隆增殖性改變,明確提示其為一種腫瘤性增生疾病[3-4]。但本病病因及發(fā)病機(jī)制迄今尚未明確,可能與免疫系統(tǒng)功能紊亂、細(xì)胞因子介導(dǎo)、酶代謝功能失調(diào)、人皰疹病毒感染等因素有關(guān)[5]。有研究認(rèn)為,朗格漢斯細(xì)胞的異常聚集和克隆性增生主要是免疫功能失調(diào)的后果,其中細(xì)胞因子和趨化因子在免疫調(diào)節(jié)中發(fā)揮了核心作用[6-7]。另有研究表明,LCH患者具有激活突變的原癌基因BRAF,由此更進(jìn)一步證明其為一種腫瘤性疾病[8]。
3.2臨床特征
LCH臨床呈多樣化表現(xiàn),是一種具有顯著異質(zhì)性特征的疾病,其既可表現(xiàn)為單一器官系統(tǒng)的孤立性病變,也可是多系統(tǒng)進(jìn)展播散性的高度惡性表現(xiàn),病情可自然緩解甚至消退亦可迅速惡化。LCH包括3種臨床綜合征:“Letterer-Siwe病”(多灶性多器官多系統(tǒng)受累,包括骨、皮膚、肝、脾和淋巴結(jié)等)、“Hand-Schuller-Christian病”(多灶性單一系統(tǒng)受累,即在同一種系統(tǒng)器官內(nèi)累及多個(gè)部位,大多為骨組織)和“孤立性嗜酸性粒細(xì)胞肉芽腫”(單一病灶,通常累及頭骨、股骨、盆骨或肋骨,少見累及淋巴結(jié)、皮膚或肺)。LCH可見于各年齡段,但兒童多發(fā),且男性患者較多見。病變可全身多部位累及,但約90%的病例發(fā)生在頭部[9]。LCH可單獨(dú)發(fā)病,亦可繼發(fā)或伴發(fā)于其他疾病[10-11]。本組6例患者均為兒童,其中4例為男性,首次發(fā)病年齡平均為7歲,4例單發(fā)生于顱面部,1例發(fā)生于小腿皮下,1例發(fā)生于全身多處皮膚。
3.3診斷
病理切片檢查是診斷LCH的最重要依據(jù),主要依靠組織形態(tài)學(xué)、免疫組化,必要時(shí)還可借助電鏡觀察。光鏡下朗格漢斯細(xì)胞彌漫增生分布,細(xì)胞中等偏大,為10~15 μm,細(xì)胞漿較少,細(xì)胞質(zhì)淡染或嗜酸性。細(xì)胞核呈園形、卵圓形、腎形或不規(guī)則形,常見核溝、折疊和凹陷。病灶背景中可見呈散在分布的嗜酸性粒細(xì)胞及多核巨細(xì)胞。免疫組化檢測(cè)腫瘤細(xì)胞幾乎一致彌漫表達(dá)CD1α和S-100,另外可不同程度表達(dá)CD68。電鏡觀察可以找到具特征性的Birbeck小體[12]。在組織病理形態(tài)學(xué)的基礎(chǔ)上,如果免疫組化CD1α和S-100陽(yáng)性則可確診LCH[13]。本組6例鏡下所見及免疫組化表型與文獻(xiàn)相同,部分病例局灶見小血管及成纖維細(xì)胞增生,病例2病灶內(nèi)可見多核巨細(xì)胞顯著增生,呈散在分布。免疫組化標(biāo)記CD68、CD1α和S-100均強(qiáng)陽(yáng)性,P53表達(dá)不明顯,Ki-67增殖指數(shù)在5%~10%之間。
3.4鑒別診斷
病理醫(yī)生在確診LCH前需要和多種腫瘤及炎癥性疾病相鑒別。①朗格漢斯細(xì)胞肉瘤:細(xì)胞異型性更加顯著,核染色質(zhì)異常豐富,核仁明顯,核分裂像多見。據(jù)文獻(xiàn)報(bào)道,每10個(gè)高倍鏡視野核分裂像計(jì)數(shù)可達(dá)30~60個(gè),病灶背景中偶見嗜酸性粒細(xì)胞浸潤(rùn)[14]。朗格漢斯細(xì)胞肉瘤免疫組化表達(dá)及電鏡同LCH。②組織細(xì)胞肉瘤:這是一種由具吞噬活性的組織細(xì)胞克隆性增生所形成的惡性腫瘤,細(xì)胞異型性明顯,免疫組化表達(dá)CD68和溶菌酶陽(yáng)性,但CD1α、S-100陰性。③反應(yīng)性竇組織細(xì)胞增生:竇組織細(xì)胞顯著增生但不具備朗格漢斯細(xì)胞的形態(tài)學(xué)特征,背景中沒有明顯的嗜酸性粒細(xì)胞浸潤(rùn),免疫組化CD68陽(yáng)性,CD1α、S-100陰性。④骨髓炎:主要以漿細(xì)胞及中性粒細(xì)胞彌漫浸潤(rùn)為主,嗜酸性粒細(xì)胞較少見,也缺乏典型的朗格漢斯細(xì)胞。⑤幼年性黃色肉芽腫:主要見于嬰幼兒,病變初期表現(xiàn)為皮膚的斑丘疹,當(dāng)杜頓型多核巨細(xì)胞及泡沫樣組織細(xì)胞不明顯時(shí)需與LCH相鑒別;臨床表現(xiàn)上患者通常無發(fā)熱、肝、脾、淋巴結(jié)腫大等系統(tǒng)性病變;鏡下組織學(xué)特征為真皮層內(nèi)組織細(xì)胞/巨噬細(xì)胞增生,核呈圓形或橢圓形,無核溝和核折疊,細(xì)胞不浸潤(rùn)表皮;免疫組化標(biāo)記CD68和lysozyme陽(yáng)性,CD1α和S-100陰性。
3.5預(yù)后
LCH患者臨床表現(xiàn)多樣,其預(yù)后差別也很大,病情可以自行緩解消退,也可導(dǎo)致死亡,病程不能通過組織學(xué)特征預(yù)測(cè)[15]。影響預(yù)后的因素主要包括發(fā)病年齡、性別、受累器官數(shù)量以及受累器官功能障礙的嚴(yán)重程度[16]。總體而言,發(fā)病年齡小、男性、累及器官多、器官功能受損嚴(yán)重、多系統(tǒng)進(jìn)展性發(fā)作、對(duì)治療缺乏反應(yīng)是最可靠的預(yù)后指標(biāo)[17]。有研究報(bào)道,多系統(tǒng)性LCH的5年生存率為23%,中位生存時(shí)間約為2.1年,病變累及到肝及造血系統(tǒng)的患者死亡率較高[18-19]。
LCH是一種少見的腫瘤性疾病,其好發(fā)人群年齡有明顯傾向性,其發(fā)病初期癥狀較隱匿且臨床表現(xiàn)多樣,因此需引起臨床醫(yī)生足夠的重視,加深對(duì)該病的認(rèn)識(shí)。對(duì)該病及時(shí)準(zhǔn)確的診斷和規(guī)范化的治療對(duì)提高患者的生存率及生存質(zhì)量起著至關(guān)重要的作用。
[1]Allen CE,Li L,Peters TL,et al.Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profilecomparedwithepidermalLangerhanscells[J]. J Immunol,2010,184(8):4557-4567.
[2]張嘉,王旖旎,王昭.朗格漢斯細(xì)胞源性腫瘤研究進(jìn)展[J].中國(guó)實(shí)驗(yàn)血液學(xué)雜志,2012,20(4):1042-1046.
[3]Hu JC,Ra S,Gutierrez MA.Cutaneous Langerhans cell histiocytosis in an elderly woman[J].Dermatol Online J,2010,16(10):6.
[4]Sayhan S,Altinel D,Erguden C,et al.Langerhans cell histiocytosis of the cervical spine in an adult:a case report[J].Turk Neurosurg,2010,20(3):409-412.
[5]Wilejto M,Abla O,Hervier B,et al.Langerhans cell histiocytosis andErdheimChesterdisease[J].CurrOpinRheumatol,2012,24(1):90-96.
[6]Garabedian L,Struyf S,Opedenakker G,et al.Langerhans cell histiocytosis:a cytokine/chemokine-mediated disorder?[J].Eur Cytokine Netw,2011,22(3):148-153.
[7]Wilejto M,Abla O,Erguden C,et al.Langerhans cell histiocytosis and Erdheim-Chester disease[J].Curr Opin Rheumatol,2012,24(1):90-96.
[8]Takahashi T,Yoshimoto M,Kondoh N.Spontaneously regressed Langerhans cell histiocytosis of lymph nodes in an elderly patient[J].Intern Med,2007,46(20):1757-1760.
[9]Régis A,Ben Salem D,Lambert A,et al.Concomitantpulmonary Langerhans cell histiocytosis and malignant lymphoma:report of two cases[J].J Radiol,2009,90(1):66-68.[10]Sapkas G,Papadakis M.Vertebral Langerhans cell histiocytosis in an adult patient:case report and review of literature[J].Am Acta Orthop Belg,2011,77(2):260-264.
[11]馬小開,王圣應(yīng).甲狀腺朗格漢斯組織細(xì)胞增生癥一例[J].中華內(nèi)分泌外科雜志,2013,7(1):79-80.
[12]許霞,劉衛(wèi)平,楊群培,等.Langerhans細(xì)胞組織細(xì)胞增生癥258例臨床病理特征及免疫表型分析[J].中華病理學(xué)雜志,2012,41(2):91-93.
[13]Dziegiel P,Dolilńska-Krajewska B,Dumańska M,et al. Coexpression of CD1a,langerin and Birbeck's granules inLangerhans cell histiocytoses(LCH)in children:ultrastructuraland immunocytochemical studies[J].Folia Histochem Cytobiol,2007,45(1):21-25.
[14]Madrigal-Martínez-Pereda C,Guerrero-Rodríguez V,Guisado-Moya B,et al.Langerhans cell histiocytosis:literature reviewand descriptive analysis of oral manifestations[J].Med OralPatol Oral Cir Bucal,2009,14(5):222-228.
[15]Ulivieri S,Oliveri G,F(xiàn)ilosomi G.Solitary Langerhans cellhistiocytosis orbital lesion:case report and review of the literature[J].Neurocirugia(Astur),2008,19(5):453-455.
[16]Ng Wing Tin S,Martin-Duverneuil N,Idbaih A,et al. Efficacy of vinblastin in central vervous system Langerhans cell histiocytosis:a nation wide retrospective study[J]. Orphanet J Rare Dis,2011,6(1):83-89.
[17]Braier J,Latella A,Balancini B,et al.Outcome in children withpulmonary Langerhans cell Histiocytosis[J]. Pediatr BloodCancer,2004,43(7):765-769.
[18]Kotecha R,Ventramani R,Jubran RF,et al.Clinical outcomes of radiantion therapy in the management of Langerhans cell Histiocytosis[J].Am J Clin Oncol,2013,28(6):365-369.
[19]Szturz P,Adam Z,Rehuk Z,et al.Lenalidomide proved effective in multisystem Langerhans cell histiocytosis[J]. Acta Oncol,2012,51(3):412-415.
Clinicopathologic analysis of 6 cases with Langerhans cell histiocytosis
QIU LiLIU YuhongWEN ShouqingWANG ChunfuCHEN LeiGONG Jinqing
Bao'an People's Hospital of Shenzhen City,Guangdong Province,Shenzhen518101,China
Objective To study the clinicopathologic characteristics,immunophenotype and prognosis of Langerhans cell histiocytosis(LCH).Methods Clinical and pathological features were studied in 6 cases of children with LCH,while the immunohistochemical staining was performed to observe the LCH immune phenotype,and combined with the literatures review.Results There were 4 males and 2 females.The youngest was 23-day-old and the oldest was 13 years.Histological changes included:diffused distribution of Langerhans cells,the cells were medium to relatively large,the nucleuses were round,oval,bean-like or folded in shape.Immunohistochemical staining showed that Langerhans cells expressed S-100,CD68 and CD1α,but been negative for LCA,CK,CD3,CD5 and CD20.Conclusion LCH presents complex clinical symptoms.The diagnosis needs a combination of clinical features,image examination and histopathological characters.The prognosis of LCH is various,depending on the age,the number of infiltratedorgans and the function of infiltrated organs.
Langerhans cell histiocytosis;Pathology analysis;Immunophenotyping
R733
A
1673-7210(2015)05(b)-0161-04
2015-01-20本文編輯:程銘)
廣東省深圳市寶安區(qū)科技計(jì)劃項(xiàng)目(2012015)。
 中國(guó)醫(yī)藥導(dǎo)報(bào)2015年14期
中國(guó)醫(yī)藥導(dǎo)報(bào)2015年14期
- 中國(guó)醫(yī)藥導(dǎo)報(bào)的其它文章
- 中文版尿毒癥期腹膜透析患者依從性問卷的開發(fā)與研究
- 以衛(wèi)生籌資視角對(duì)非洲國(guó)家實(shí)現(xiàn)全民健康覆蓋的進(jìn)展與挑戰(zhàn)的探討
- 延續(xù)護(hù)理對(duì)糖尿病患者血壓、血糖以及血脂水平變化的影響
- 流程責(zé)任小組對(duì)手術(shù)室護(hù)理服務(wù)水平的影響
- 舒適護(hù)理結(jié)合生命力保養(yǎng)在骨外傷患者中的應(yīng)用效果
- 優(yōu)質(zhì)護(hù)理理念在子宮內(nèi)膜息肉宮腔鏡術(shù)后患者中的應(yīng)用價(jià)值