脊髓性肌萎縮癥2個核心家系SMN1基因分析
2015-10-22 12:44:32曾光群楊季云張丁丁通訊作者
中國實用神經疾病雜志 2015年23期
關鍵詞:檢測
曾光群 楊季云 張丁丁(通訊作者) 陳 蓉 李 寧
1)四川彭州市人民醫院 彭州 611930 2)四川省醫學科學院 四川省人民醫院人類疾病基因研究四川省重點實驗室 成都 610072 3)川北醫學院 南充 637000 4)遵義醫學院 遵義 563000
脊髓肌肉萎縮癥(spinal muscular atrophy,SMA)是僅次于囊性纖維化的兒童致死性疾病,是由于脊髓前角運動神經元變性引起的漸進性近端肌肉無力和癱瘓的神經肌肉性疾病,大多數為常染色體隱性遺傳,也有報道為常染色體顯性遺傳和X 連鎖遺傳[1]。發病率1/6 000~1/10 000,攜帶率在不同的人群中為1/40~1/60[2-3],我國南部正常人群此病的攜帶率約1/60[4],上海、臺灣正常人群此病的攜帶率約1/39[5]和1/48[6]。
1995年法國學者將SMA 的基因定位于5q11.2~13.3,認為反向重復的運動神經元生存基因(survival motor neuron gene,SMN)為致病基因[7]。該基因有2個序列高度相似的拷貝:靠近端粒的決定性基因SMN1和靠近著絲粒的修飾基因SMN2,兩者間有5個堿基的區別,編碼序列內僅1 個堿基差異。SMN 所在的染色體區域內結構復雜,且存在眾多重復序列和假基因簇,致其結構不穩定,發生缺失或轉換的頻率較高,使相應的SMN1基因拷貝數復雜多變。95%以上的SMA 患者有SMN1第7號外顯子純合缺失,其余5%是SMN1點突變或者復合型的雜合缺失[1]。SMN2 基因的拷貝數是SMN1基因缺失的劑量補償,與患兒臨床表型的嚴重程度相關[8]。MLPA 是一種檢測基因缺失或重復的高效、準確方法[9],STR 連鎖能分析風險染色體的來源。本研究聯合兩種方法對2個SMA 家系成員SMN1基因檢測,明確了基因攜帶情況,給患兒和家庭提供了完整的評估。
1 資料和方法
1.1 一般資料 先證者1,女,11歲,G1P1,足月順產,圍生期無異常。3歲前運動無異常,自3歲開始出現下蹲困難,易摔等現象,下肢比上肢嚴重。體格檢查:身高130cm,四肢肌肉萎縮,肌力、肌張力及膝腱反射減弱,病理征未引出。頭顱MRI未見明顯異常,肌電圖表現為神經源性損害。輔助檢查:磷酸肌酸激酶324U/L(參考值26.00~174.00U/L),磷酸肌酸激酶同工酶52U/L(參考值0~25.00U/L)。患兒父母體健,非近親結婚,家族中無同類病史。
先證者2,男4歲。G1P1,足月順產,圍生期無異常。患兒自幼運動發育落后,2歲左右開始出現運動能力倒退,走路不穩,上下樓梯困難,下蹲后不易站起,呈鴨子步態。體格檢查:體型瘦小,雙側腓腸肌略肥大,四肢肌力Ⅳ級、肌張力降低,膝腱反射減弱,病理征陰性,Gower征(+)。輔助檢查:磷酸肌酸激酶408U/L,磷酸肌酸激酶同工酶62U/L,肌電圖呈典型的失神經性改變。患兒父母體健,非近親結婚,家族中無同類病史,母親懷孕18 周。
1.2 研究方法
1.2.1 基因組DNA 提取:簽署知情同意書后,取患兒及家人乙二胺四乙酸鈉抗凝外周血2 mL,無菌抽取孕婦羊水5 mL。用DNA 抽提試劑盒(北京天根生化科技有限公司)提取基因組DNA,操作步驟嚴格按照試劑盒說明書進行。NanoDrop2000測定濃度。將DNA 濃度校正至50ng/μL,-20 ℃保存。
1.2.2 MLPA 檢測:按荷蘭MRC-Holland公司提供的MLPA 試劑盒檢測。DNA 樣本(50ng)5μL,經變性、探針雜交過夜、連接和PCR 反應等步驟后,取PCR 產物1.0μL,加1.5μL LIZ-500,HD 7μL,混勻,ABI3130xl測序儀進行片段分析[10]。
1.2.3 結果判定:使用GeneMapper軟件收集數據,Coffalyser軟件進行分析。如無信號則為外顯子缺失,表現為相關峰消失,信號成倍增加表示發生重復。攜帶者相應外顯子缺失信號降低35%~55%。
1.2.4 STR 基 因 連 鎖 分 析:根 據 文 獻[11],選 用4 個 連 鎖STR 位點(D5S435、D5S351、D5S610、D5S629)對2個家系分析。引物由上海生工合成,熒光標記引物兩翼,ABI3130xl遺傳分析儀上機檢測,進行單倍型連鎖分析。
2 結果
2.1 MLPA 結果 家系1中先證者SMN1基因第7號外顯子及8號外顯子拷貝數均為零,母親和外祖母SMN1基因第7號外顯子及8號外顯子為1個拷貝數,其爺爺SMN1基因第7號外顯子及8號外顯子為3個拷貝數。父親SMN1基因第7號外顯子及8號外顯子為2個拷貝數。家系2先證者SMN1基因第7號外顯子及8號外顯子拷貝數均為零,母親和胎兒SMN1基因第7號外顯子及8號外顯子為1個拷貝數,父親SMN1基因第7號外顯子及8號外顯子為2個拷貝數。
2.2 單倍型分析結果 家系1先證者與其父親有相同的母源致病的SMN1等位基因。家系2 先證者和胎兒均遺傳1條父親的致病的等位基因片段。
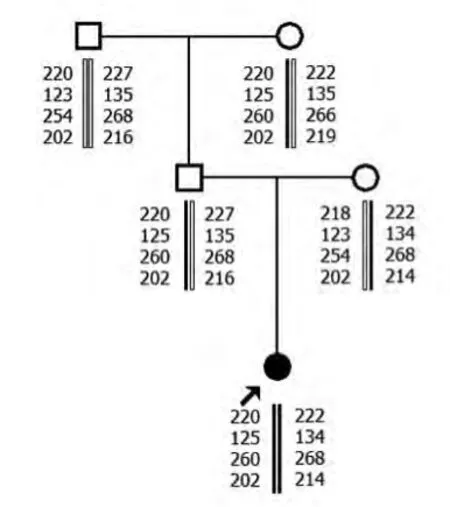
家系1
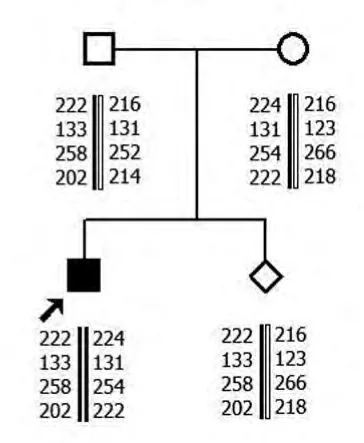
家系2
3 討論
SMA 的診斷主要依靠臨床表現、實驗室檢查、家族遺傳史及基因檢測4個方面,但臨床表現和常規輔助檢測缺乏特異性,故臨床上對確診SMA 更多依賴于基因檢測。臨床上SMA 各型間的差異很大,依據發病年齡和臨床病程嚴重程度的不同,分為Ⅰ型、Ⅱ型、Ⅲ型和Ⅳ型,其中Ⅰ型約占70%,臨 床 表 型 最 嚴 重,多 數 于6 個 月 內 起 病,2 歲 內 夭 折[12]。SMN1基因缺失(包括SMN1基因純合缺失或SMN1基因轉換為SMN2基因)或SMN1基因內微小突變是引起SMA 的主要原因。SMN1基因缺失及其附近基因的大范圍缺失,引起的臨床表型較嚴重,SMN1基因轉換為SMN2基因,引起的臨床表型較輕,但SMN 基因的缺失和表型的嚴重程度缺失之間沒有相關性[13]。
每條染色體上有0~4個拷貝的SMN 基因,大部分純合缺失的患者SMN1拷貝數為0,健康人SMN1拷貝數為2,攜帶者和雜合缺失的患者SMNI拷貝數為1,約5%的正常個體可有3~4個SMN1拷貝,曲曉星等[5]報道在正常個體中有5~7個拷貝。因而通過檢測SMN1拷貝數可診斷SMA患者以及篩查攜帶者。SMA 攜帶者主要有4種基因型[14]:2條同源染色體上,1 條染色體上有1 拷貝正常的SMN1 基因,另1條缺失SMN1基因的為“1+0”型;1條染色體上有2拷貝SMN1基因,另1條缺失SMNI基因的“2+0”型;1條染色體上有1拷貝SMN1基因,另1條SMN1基因發生微小突變的“1+1m”型;1條染色體上有2拷貝SMNI基因,另1條SMN1基因發生突變的“2+1m”型;“1+0”型攜帶者最常見,約占95%,其他約占5%。
本研究先證者1和2SMN1基因的第7號外顯子及8號外顯子拷貝數均為零,可在基因水平診斷先證者為SMA。先證者1母親和外祖母SMN1基因第7號外顯子及8號外顯子為1個拷貝數,先證者2母親SMN1基因第7號外顯子及第8號外顯子為1個拷貝數,為雜合缺失突變,提示其均為攜帶者。先證者1爺爺SMN1 基因第7 號外顯子及第8號外顯子為3個拷貝數,SMN1基因正常。先證者1和2父親SMN1基因第7號外顯子及8號外顯子為2個拷貝數,表型上為正常。但其家中卻有患兒出現,結合STR 分析,顯示先證者遺傳了1條父親致病的等位基因片段,為“2+0”SMA致病基因攜帶者,即1條染色體上有2拷貝SMN1基因,另1條染色體缺失SMN1基因。家系2胎兒SMN1基因第7號外顯子及8號外顯子為1個拷貝數,1條為父源致病等位基因片段,1條為母源正常等位基因片段,為SMA 攜帶者,可以生育。
SMA 的嚴重程度由兩條染色體上攜帶突變的SMN 基因、SMN2基因拷貝數等其他因素決定。當前檢測SMA 攜帶者的方法,尚存在一定局限性。“1+1m”型和“2+0”型的SMA 攜帶者每條染色體上含SMN1相同劑量的拷貝數,因此普通人群中檢測出含有2拷貝SMN1只能說明攜帶者的風險降低,不能完全排除“1+1 m”型雜合缺失和“2+0”型SMA 攜帶者,在下一代中仍可有發病者。由于SMA 是一種發病率較高的遺傳性致死性疾病,基因攜帶率大,至今尚無有效的治療措施,產前診斷是預防該病的有效手段,因而患者的診斷以及攜帶者的檢出尤為重要。我們聯合MLPA 和STR 連鎖分析方法對2個核心家系SMN1基因分析,驗證了先證者的風險等位基因的來源,檢出有2個拷貝的“2+0”型SMA 攜帶者,增加了檢出率。
通過以上分析,我們發現將MLPA 和STR 連鎖分析的聯合應用,可充分發揮二者的優勢,明確SMA 攜帶者,確診先證者。臨床上考慮為SMA 患者應對以下情況進行SMN檢測:(1)有SMA 生育史;(2)自身為SMA 攜帶者,欲生育,則須對其配偶進行攜帶者檢測;(3)夫妻雙方家族均有脊髓性肌肉萎縮癥病史;(4)夫妻均為攜帶者或曾經是脊髓性肌肉萎縮癥患者;(5)先證者經基因診斷明確的高危產婦的。為保證產前診斷的準確性,應結合多態性連鎖分析以減少風險發生率。對于無SMN1 純合缺失的患者,對其基因內微小突變檢測。對基因確診的患者,要分析5q13 區域內其他修飾基因的變異,預測臨床表型和病情進展。
[1] Jiang W,Ji X,Xu Y,et al.Molecular prenatal diagnosis of autosomal recessive spinal muscular atrophies using quantification polymerase chain reaction[J].Genet Test Mol Biomarkers,2013,17(5):438-442.
[2] Markowitz JA,Singh P,Darras BT.Spinal muscular atrophy:a clinical and research update[J].Pediatr Neurol,2012,46(1):1-12.
[3] Kocheva SA,Plaseska-Karanfilska D,Trivodalieva S,et al.Prenatal diagnosis of spinal muscular atrophy in Macedonian families[J].Genet Test,2008,12(3):391-393.
[4] Prior TW.Spinal muscular atrophy diagnostics[J].J Child Neurol,2007,22(8):952-956.
[5] 曲曉星,肖冰,季星,等.應用熒光定量PCR 開展上海地區脊肌萎縮癥攜帶者的人群篩查[J].中華醫學遺傳學雜志,2013,30(1):1-4.
[6] Su YN,Huang CC,Lin SY,et al.Carrier screening for spinal muscular atrophy(SMA)in107,611pregnant women during the Period 2005-2009:A prospective population-based cohort study[J].PloS One,2011,6(2):e17 067.
[7] Roy N,McLean MD,Besner-Johnston A,et al.Refined physical map of the spinal muscular atrophy gene(SMA)region at 5q13based on YAC and cosmid contiguous arrays[J].Genomics,1995,26(3):451-460.
[8] 盧麗萍,麻宏偉,姜俊,等.脊髓型肌萎縮臨床表型與基因拷貝數變化的相關性研究[J].中華遺傳學雜志,2007,24(2):144-147.
[9] Lalic T,Vossen RH,Coffa J,et al.Deletion and duplication screening in the DMD gene using MLPA[J].Eur J Hum Genet,2005,13(11):1 231-1 234.
[10] Schouten JP,McElgunn CJ,Waaijer R,et al.Relative quantification of 40nucleic acid sequences by multiplex ligation-dependent probe amplification[J].Nucleic Acids Res,2002,30(12):e57.
[11] 孫維,沈嘉瑋,龍飛,等.優選短串聯重復序列應用于脊髓肌萎縮癥產前診斷的連鎖分析[J].上海交通大學學報,2010,30(6):707-712.
[12] Zerres K,Rudnik-Schoneborn S.Natural history in proximal spinal muscular atrophy.Clinical analysis of 445patients and suggestions for a modification of existing classifications[J].Arch Neurol,1995,52(5):518-523.
[13] Dastur RS,Gaitonde PS,Khadilkar SV,et al.Correlation between deletion patterns of SMN and NAIP genes and the clinical features of spinal muscular atrophy in Indian patients[J].Neurol India,2006,54(3):255-259.
[14] Chen Wj,Wu ZY,Wang N,et al.Quantitative studies on SMN1gene and carrier testing of Spinal muscular atrophy[J].Chin J Med Genet,2005,22(6):599-602.
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
