基于改進隱馬爾科夫模型的畜禽全基因組關聯分析中的多重檢驗方法
2015-06-15 01:14:26梅步俊王志華
安徽農學通報 2015年10期
梅步俊 王志華

摘 要:為了改進在畜禽全基因組關聯分析中,利用隱馬爾可夫模型(HMM)進行多重檢驗時的過學習問題,提出將人工神經網絡算法(ANN)作為預處理,引入畜禽全基因組關聯分析中,較好地彌補了已有的多重檢驗方法的缺陷,提高了統計推斷性能,其運算速度也顯著提高。
關鍵詞:全基因組關聯分析;隱馬爾科夫模型;人工神經網絡;多重比較;假設檢驗
中圖分類號 S852 文獻標識碼 A 文章編號 1007-7731(2015)10-22-03
Abstract:In order to improve the learning problem of hidden Markov Model(HMM)for multiple testing in whole-genome Association analysis of livestock and poultry,a algorithm of artificial neural network(ANN)as a pretreatment is proposed in multiple testing methods of genome-wide association analysis of livestock and poultry. Results showed that it just well make up for the deficiencies of multiple testing methods based on HMM,and improve the performance of statistical inference,and its speed is also improved significantly.
Key words:Genome-wide association study(GWAS);Hidden Markov Model(HMM);Artificial neural network(ANN):Multiple comparisons; Hypothesis testing
全基因組關聯分析(Genome-Wide Association Studies,GWAS)是基于“常見疾病,常見變異(common disease,common variant)”的假設,利用標記和突變位點在群體水平的連鎖不平衡檢測QTL。這種關聯性的產生是由于當前群體攜帶有源于共同祖先的染色體片段,這些片段包含相同的QTL等位基因或單倍型。一般認為,對于復雜性狀,除了受少數幾個效應較大的基因控制外,還受許多微、中效基因控制。隨著對基因組信息認識水平的的提高和高通量測序技術的發展,GWAS已經變成研究復雜性狀遺傳機理的重要手段。截至2014年7月,僅在人類上就有1 927篇GWAS文章發表,共報道13 418個SNP與各類性狀有顯著性相關。國內外不少研究者對畜禽的重要經濟性狀、遺傳缺陷性疾病、復雜疾病的抗性、品種特征等性狀也開展了GWAS。Zhang等[1]使用澳大利亞996頭婆羅門牛和1 097頭有6次產犢記錄的母牛進行繁殖性狀的遺傳評估。Santana等[2]使用720頭公瘤牛的平均日增重(ADG)和354 147SNP數據進行GWAS,采用混合模型和回歸方法檢測到3號染色體上的6個SNP與ADG顯著相關。顯著性最高的SNP(p=9.49×10-8)解釋了5.62%的表型方差。
大尺度多重檢驗(large scale multiple test)是現代統計學的重要研究領域,廣泛應用在GWAS、DNA芯片分析和腦圖像分析等領域。在這些研究中,常常同時檢測數以萬計甚至百萬假設檢驗。在GWAS中,常常使用Bonferroni矯正多重比較問題,但是由于連鎖不平衡,SNP之間是不獨立的,這與傳統假設檢驗所有假設之間獨立、所有樣本來源于單一分布不同。例如,不同的基因可能在同一通路中,這些基因表現較強的相關性。畜禽群體中,由于個體之間存在親緣關系和共同環境效應,這都會對多重檢驗造成顯著影響。Finner和Roters[3]、Owen[4]研究表明假設檢驗間的相關性顯著影響Ⅰ型錯誤率的期望和方差。Sun和Tony等[5]應用復合決策理論,由隱馬爾科夫模型(HMM)研究假設檢驗間不獨立情況下的多重檢驗問題,構建“局部顯著性指數”(local index of significance,LIS)替代p值,改進了假設檢驗間不獨立情況下的假發現率(false discovery rate,FDR)性能。Wei和Sun等[6]應用類似的方法發展了“匯集局部顯著性指數”(pooled local index of significance,PLIS)控制FDR,該方法具有最小的假陰性率(false negative rate,FNR),比傳統的基于P值的假設檢驗方法有更高的功效。Li和Wei等[7]利用SNP間的LD信息作為先驗信息,建立加權LD圖模型,應用馬爾科夫隨機場模型(Markov random field model,HMRF)控制GWAS中的假發現率。但基于單倍型的方法可能由于自由度太高、單倍型推斷中的錯誤和單倍型包含SNP數的武斷性而損失功效。現有基于HMM的多重檢驗方法存在過學習的問題,訓練出的模型可能存在過擬合情況。本研究將人工神經網絡算法(ANN)和HMM結合,ANN算法具有很強的抗干擾性,也不存在過學習的問題,彌補了HMM的缺陷,提高了多重檢驗的功效。
1 研究背景
1.1 隱馬爾可夫模型 設[θ=θm1=θ1,…,θm]是服從伯努利分布的隨機變量,[θi=0]表明變量[i]來源于零假設,反之來源于非零假設。假設觀察值[x=x1,…,xm]由以下條件概率產生:
1.3 人工神經網絡算法(ANN)人工神經網絡是一種應用類似于大腦神經突觸聯接的結構進行信息處理的數學模型。神經網絡是一種運算模型,由大量的節點(或稱神經元)之間相互聯接構成。每個節點代表一種特定的輸出函數,稱為激勵函數(activation function)。每2個節點間的連接都代表一個對于通過該連接信號的加權值,稱之為權重,這相當于人工神經網絡的記憶。網絡的輸出則依網絡的連接方式,權重值和激勵函數的不同而不同。鑒于ANN和HMM的互補性,利用ANN的長處來克服HMM的不足,將ANN作為HMM的預處理部分,利用ANN的抗干擾能力改進HMM的過學習,具有預測精度高、耗時少的特點。
2 材料與方法
2.1 第15屆QTL-MAS公共數據集 該公共數據由一個遠交群體構成,使用 LDSO 軟件[9]模擬產生。歷史群體首先模擬了1 000個世代,每個世代1 000個個體,隨后模擬30個世代,每個世代150個個體。基因組共模擬了5條染色體、9 990個 SNP標記,每條染色體長度1M(Morgan),攜帶均勻分布的SNP標記1 998個(相鄰SNP標記間距0.05cM)。最終用于基因組選擇評估的數據由3 220個個體組成,其中包括20頭公畜、200頭母畜(每頭公畜與10頭母畜交配)和3 000個后裔(每頭母畜生產 15 個后裔)。所有個體都模擬了9 990個SNP 標記的基因型,并且沒有基因型缺失或判型錯誤。每頭母畜的15個后裔中,有10個模擬了一個連續性狀的表型值。2 000個有表型值的后裔及其它1 000個沒有表型值(但有模擬的真實育種值)的后裔,分別作為參考群和驗證群。
2.2 模擬數據 全基因組數據模擬需要首先定義基因組的結構。與基因組結構相關的參數包括:染色體長度(Lc)、染色體數(Nc)、總標記數(Nm)、標記位置的分布和基因數或數量性狀基因座(QTL)數(NQTL)。為便于模擬,一般設定不同染色體的長度相同,為1M(Morgan),不同染色體上的標記數目一般也假設相同。
2.3 模擬研究 結合HMM和ANN,利用ANN彌補HMM的不足。HMM訓練模型主要基于最大似然準則(Maximum Likelihood,簡稱ML)和最大共有信息準則(MaximumMutual Information,簡寫為MMI),采用梯度法計算,更新HMM模型中的參數。采用最小均方誤差(MMSE)準則,ANN提高HMM的識別能力,保證H0,H1推斷的正確性。具體步驟:建立3層ANN結構(一個輸入層,一個隱含層,一個輸出層)和隨機初始ANN神經元鏈接權重,由n個輸入節點和n個輸出節點組成。采用類似于交叉驗證策略,將模擬數據真實值作為輸入,訓練鏈接權重。將ANN算法的輸出,輸入到HMM模型中,采用Baum-Welch算法得出對數似然值,應用梯度下降法調整模型參數。測試的誤差評價準則為平均相對誤差:
3 結果與分析
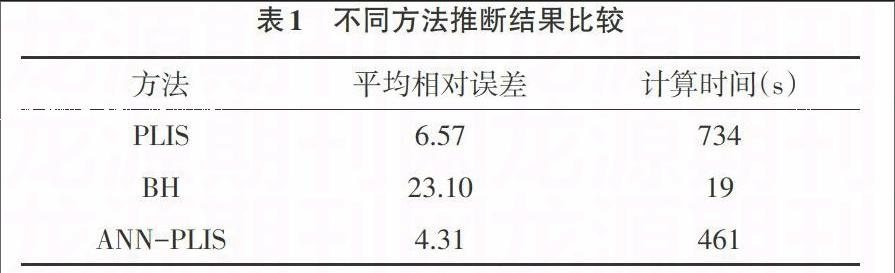
研究采用自編軟件產生模擬數據,結合公共數據集,將ANN和HMM相結合,提出改進PLIS法-ANN-PLIS。通過比較PLIS、BH[11]、ANN-PLIS的預測性能,結果表明,ANN-PLIS較單純使用HMM的PLIS運算速度很快,ANN-PLIS具有很強的魯棒性、記憶能力、非線性映射能力以及強大的自學習能力,運算速度也大大加快,提高了多重比較過程的準確性(表1)。但是ANN也有其缺點:(1)最嚴重的問題是沒能力來解釋自己的推理過程和推理依據;(2)不能向用戶提出必要的詢問,而且當數據不充分的時候,神經網絡就無法進行工作;(3)把一切問題的特征都變為數字,把一切推理都變為數值計算,其結果勢必是丟失信息;(4)理論和學習算法還有待于進一步完善和提高。
致謝:本研究部分靈感及部分計算設備由中國農業大學動物科技學院張勤教授課題組提供。
參考文獻
[1]Zhang,Y.D.,et al.. Genomic selection for female reproduction in Australian tropically adapted beef cattle[J]. Animal Production Science,2014,54(1):16-24.
[2]Santana,M.H.,et al.,Genome-wide association study for feedlot average daily gain in Nellore cattle(Bos indicus)[J]. J Anim Breed Genet,2014,131(3):210-216.
[3]Finner,H.,M. Roters. Multiple hypotheses testing and expected number of type I[J].Ann. Statist.,2002:220-238.
[4]Owen,A..Variance of the number of false discoveries[J].Journal of the Royal Statistical Society 2005,B(67):411-426.
[5]Sun,W.,T. Tony Cai. Large-scale multiple testing under dependence[J]. Journal of the Royal Statistical Society:Series B(Statistical Methodology),2009,71(2):393-424.
[6]Wei,Z.,et al..Multiple testing in genome-wide association studies via hidden Markov models[J].Bioinformatics,2009,25(21):2802-2808.
[7]Li,H.,Z. Wei,J. Maris. A hidden Markov random field model for genome-wide association studies[J]. Biostatistics,2010,11(1):139-150.
[8]Wenguang,S.,T.T. Cai,Large-scale multiple testing under dependence[J]. Journal of the Royal Statistical Society Series B,2009,71(2):393-424.
[9]Ytournel,F..Linkage disequilibrium and QTL fine mapping in a selected population[J].Station de Génétique Quantitative et Appliquée,INRA,2008.
[10]Meuwissen,T.,M. Goddard.Accurate Prediction of Genetic Values for Complex Traits by Whole Genome Resequencing[J].Genetics,2010,185:623-631.
[11]Benjamini,Y.,Y. Hochberg.Controlling the False Discovery Rate:A Practical and Powerful Approach to Multiple Testing[J].Journal of the Royal Statistical Society. Series B(Methodological),1995,57(1):289-300. (責編:張宏民)
