2-亞氨基-1,2-二氫吡啶-1-乙酸的合成
2015-05-10 08:00:50石桂珍由文穎張兆貴孫建梅
天津化工 2015年6期
關鍵詞:振動
石桂珍,由文穎,張兆貴,孫建梅
(濰坊工程職業學院應用化學與生物工程學院,山東青州 262500)
2-亞氨基-1,2-二氫吡啶-1-乙酸則是合成咪唑黃隆的重要中間體,由于除草劑抗性問題的存在,新的除草劑中間體的設計和合成仍是農業化學中一個持續的挑戰,日本專利[5]及劉長令[6]報道了該中間體的合成,但對其實驗條件未作深入探索。本文在此基礎上對實驗條件進行了探索,優化了反應條件,以期對其工業化生產提供可利用的數據。
1 實驗部分
1.1 試劑及儀器
實驗中所用氯乙酸、無水乙醇和三乙胺均為分析純試劑,2-氨基吡啶為化學純試劑。
所用儀器包括X-6顯微熔點測定儀、Finigan LCQ Advantage型質譜儀、PE-2400型元素分析儀(美國PEKIN-ELMER公司)、Varian INOVA 400 M NMR超導核磁共振波譜儀(美國Varian公司)、AVATAR 360型傅立葉變換紅外分光光度計(固體KBr壓片)及N-1001型旋轉蒸發儀等。
1.2 反應原理
2-氨基吡啶在水和乙醇作溶劑,三乙胺作縛酸劑的條件下,與氯乙酸作用生成2-亞氨基-1,2-二氫吡啶-1-乙酸。
由于氯的電負性較大,而羰基又是吸電子基,所以氯乙酸中的α碳帶部分正電荷,該碳原子與吡啶環上具有孤對電子的氮相互吸引形成中間過渡態,最終氯乙酸中的氯奪取氨基上的氫形成氯化氫離去,氨基吡啶發生雙鍵重排得亞胺結構。
1.3 實驗過程
將氯乙酸(12.5 g)溶于20 mL水和5 mL無水乙醇的混合溶液中,冷卻攪拌下在10~15℃下加入18.5 mL三乙胺,撤去冷卻裝置,隨后加入2-氨基吡啶12.5 g。將反應液加熱到75~80℃回流反應5 h后(隨著反應的進行,反應液顏色不斷加深,由無色到橙紅色再到磚紅色),向其中加入25 mL無水乙醇(反應液出現分層:上層紫紅色,下層橙色沉淀),冰水浴冷卻后過濾,并用乙醇(3×20 mL)洗滌濾餅,干燥得白色針狀固體16.0 g,收率79.2%(文獻值80%[5]),熔點249.5~251.1 ℃(文獻值250 ℃[5])。
Anal.(%)C7H8N2O2,found(calcd)C:55.13(55.26),H:5.37(5.26),N:18.33(18.42)。
2 結果與討論
2.1 FTIR分析

圖1 2-亞氨基-1,2-二氫吡啶-1-乙酸的FTIR圖
由圖1可以看出:3424 cm-1處吸收為胺類N—H伸縮振動;3246 cm-1處吸收為羧酸O—H伸縮振動;3042 cm-1處吸收為芳環C—H伸縮振動;2872 cm-1處吸收為烷烴C—H伸縮振動;1697 cm-1處吸收為羧酸C—O伸縮振動;1649 cm-1處吸收為N—C—C—C共扼伸縮振動;1627 cm-1處吸收為亞胺N—H變形振動;1586、1524 cm-1處吸收為芳環C—C伸縮振動,780 cm-1處吸收為芳環—C—H的變形振動(面外);1368 cm-1處吸收為—CH2—(—CH2—COO—)變形振動;1329 cm-1處吸收為C—N伸縮振動;1293 cm-1處吸收為羧酸C—O伸縮振動與O—H變形振動偶合。與2-氨基吡啶的紅外譜圖相比[7],其伯胺基3300~3500 cm-1處的兩個中強吸收峰轉變為3424 cm-1處仲胺的一個中強吸收峰;產物中多了3246 cm-1處羧酸的O—H伸縮振動吸收峰。
2.2 MS分析
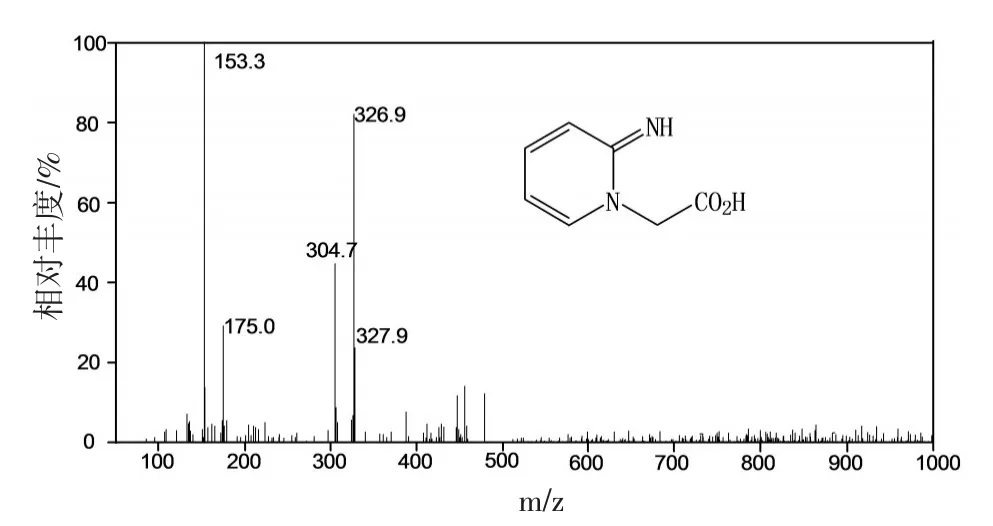
圖2 2-亞氨基-1,2-二氫吡啶-1-乙酸的MS圖
圖2為2-亞氨基-1,2-二氫吡啶-1-乙酸的MS圖,由于產物的相對分子質量為152,故MS圖中的基峰(153.3)為M+1峰。
2.3HNMR分析
以TMS為內標采用Varian INOVA 400 M NMR超導核磁共振波譜儀(美國Varian公司)測得2-亞氨基-1,2-二氫吡啶-1-乙酸的化學位移值為:1H NMR(400 MHz,D2O)4.55(s,2H),6.70(t,J=15.2,1H),6.86(d,J=8.4,1H),7.55(d,J=7.6,1H),7.63(t,J=15.2,1H)。
3 反應條件對產物收率的影響
3.1 反應時間對2-亞氨基-1,2-二氫吡啶-1-乙酸收率的因素
由圖3可見,隨著反應時間的增加,該反應的收率上升較快,反應2 h收率達到56.3%,4~5 h后收率增加不明顯,因此該反應的適宜反應時間為4~5 h。
3.2 反應溫度對2-亞氨基-1,2-二氫吡啶-1-乙酸收率的因素
由圖4可見,隨溫度升高,收率呈上升趨勢,并且溫度越高,收率變化越明顯,故該反應的適宜反應溫度為該體系的極限溫度:75~78℃,此時反應液處于沸騰狀態。主要是因為該步反應生成固體,沸騰能夠使兩相更充分接觸,有利于提高產物收率。
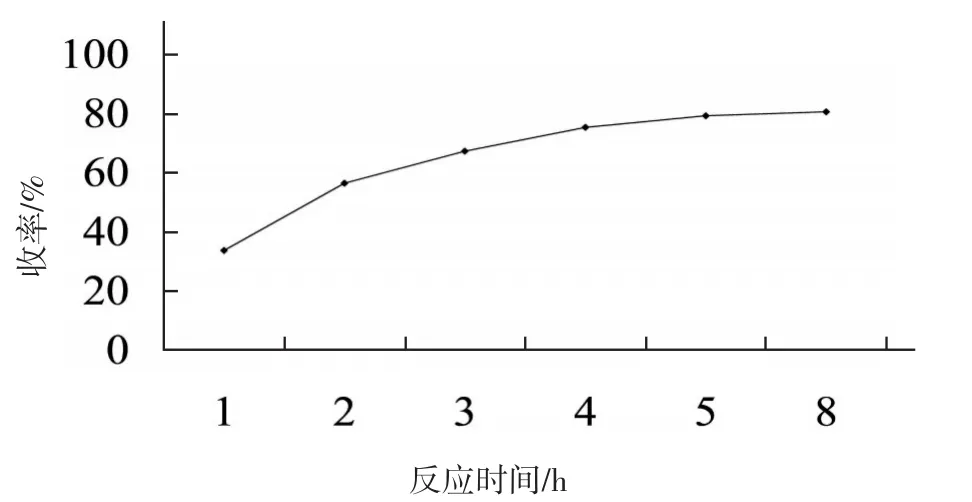
圖3 反應時間對收率的影響
此外,作者還考察了縛酸劑對產物收率的影響。縛酸劑即捕獲酸的試劑,在許多化學農藥的合成反應中,如果產物有酸生成又不便及時除去,可加入少量縛酸劑(大部分可回收),來大大提高反應速度和產物轉化率[8]。在氨基吡啶與氯乙酸反應的過程中要釋放氯化氫,而原料中又有游離的氨基存在,會發生如下副反應:
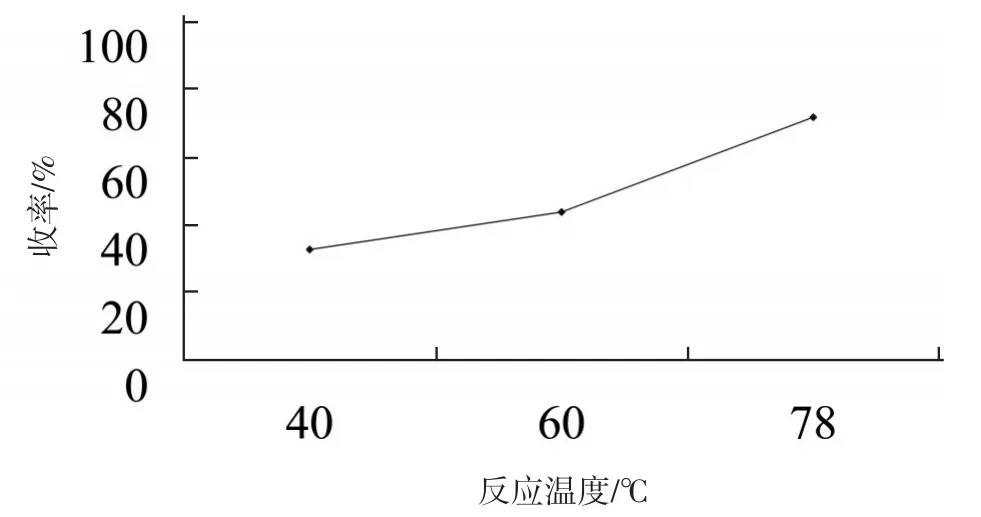
圖4 反應溫度對收率的影響

此反應的發生降低了原料的反應活性。因此,在這類反應中一般都要加入一定量的縛酸劑來中和反應生成的氯化氫。常用的縛酸劑有三乙胺、N,N-二甲基苯胺、吡啶等,本實驗選擇三乙胺為縛酸劑。通過實驗比較,不加縛酸劑時副產物明顯多于加縛酸劑時的副產物,且產物的顏色較深。不加縛酸劑時收率只有36%左右,加縛酸劑時收率可達70%以上。
3.3 正交試驗優選
由前面的探索實驗結果,本文選取反應溫度(A)為60、70、78 ℃,反應時間(B)分別為2、4、6 h,投料比(C,即氯乙酸與氨基吡啶的物質的量之比)分別為1、1.2、1.5,以乙醇和水作溶劑,加入三乙胺作縛酸劑,正交實驗結果如表1所示。

表1 正交實驗結果與極差(R)分析
3.3.1 直觀分析
極差R的大小反映了因子影響指標的主次關系,由直觀分析可以看出,影響該步合成反應收率的主次順序為:A>B>C;并得到最優實驗條件為:反應時間為6 h,投料比為1.5,反應溫度為78℃。
3.3.2 方差分析
直觀分析不能給出誤差的大小,也就不知道結果的精度,而方差檢驗能夠反應數據的波動性,即數據的分散性,方差大小表明數據變化的顯著程度,也表明因素對指標影響的大小。氯乙酸與氨基吡啶的物質的量之比對反應收率的影響較小,可并入誤差項,計算各均方及F值,列方差分析表見表2。

表2 方差分析
經方差分析可知,A、B因素對收率的影響均很顯著,且顯著性次序為A>B。結合生產實際,從降低成本、節約時間等方面考慮,可取適宜的水平為A3B2C1。
在選取的適宜水平下(反應時間為4 h,反應溫度為78℃,氯乙酸與氨基吡啶的物質的量比為1:1)做驗證實驗,所得產品收率為79.0%,收率較高,且降低了原料成本。
4 結論
4.1 實驗制得的2-亞氨基-1,2-二氫吡啶-1-乙酸經分析和表征,與預期結果相符。
4.2 影響2-亞氨基-1,2-二氫吡啶-1-乙酸收率的因素有反應時間、反應溫度及縛酸劑等。通過單因素試驗、正交試驗及方差分析,并結合工業化生產實際,可選取適宜的反應時間為4 h,適宜的反應溫度為78℃,氯乙酸與氨基吡啶的物質的量之比為1:1,乙醇和水作溶劑,加入三乙胺作縛酸劑。
4.3 對反應條件的探索為大規模工業化生產提供了可以參考的數據。
[1]TATSUO N,KAZUNARI O,SHIGEYUKI I,et al.Production of condensed heterocyclic rings:JP,1316379[P],1989-12-21.
[2]劉長令.新型稻田除草劑咪唑黃隆[J].農藥,1995,34(9):27-30.
[3]闞錦晴,李想,李永舫.聚-2-氨基吡啶電化學合成及性質[J].物理化學學報,2002,18(2):106-111.
[4]劉建超,陳偉志,賀紅武.農藥化學中的綠色化學[J].化學通報,2004,(10):750-755.
猜你喜歡
科學大眾(2023年17期)2023-10-26 07:39:14
大電機技術(2022年5期)2022-11-17 08:12:48
天天愛科學(2020年6期)2020-09-10 07:22:44
瘋狂英語·新讀寫(2020年3期)2020-06-06 09:05:56
數學物理學報(2018年4期)2018-09-14 03:40:58
數學物理學報(2017年6期)2018-01-22 02:26:40
船海工程(2015年4期)2016-01-05 15:53:26
噪聲與振動控制(2015年4期)2015-01-01 07:08:44
計算物理(2014年2期)2014-03-11 17:01:44
鄭州大學學報(理學版)(2014年3期)2014-03-01 04:21:00
