美國藥品審評質量管理規范評介及對我國的啟示
2015-03-11 07:07:53耿曉雅
中國衛生政策研究 2015年2期
耿曉雅 邵 蓉
中國藥科大學國際醫藥商學院 江蘇南京 211198
?
美國藥品審評質量管理規范評介及對我國的啟示
耿曉雅*邵 蓉
中國藥科大學國際醫藥商學院 江蘇南京 211198
本文采用文獻研究法,在概述美國藥品審評質量管理規范的政策背景、發展歷史和現狀的基礎上,重點介紹其為實現政策目標而采取的相應措施,并以1993—2013年美國藥品審評時間中位數和首輪藥品批準率數據來說明其實施效果。研究認為,一套科學完善的藥品審評質量管理規范可有效保證藥品審評質量、提高藥品審評效率。然而目前我國藥品審評質量管理規范還不完善,建議通過細化我國藥品審評時限規定、制定可操作性強的審評模板、重視藥品審評質量管理規范體系的相關培訓和持續改進等措施進一步完善我國藥品審評質量管理規范。
美國; 藥品審評; 藥品審評質量管理規范
藥品作為一種特殊商品,其安全、有效、質量可控與公眾健康密切相關,各國政府均對其實行嚴格的監管措施。[1]其中,藥品上市前的技術審評結論將作為后續監管措施的基礎和依據,其科學、高效與否關系到公眾用藥的安全性和可及性。[2]因此,藥品注冊和相應的技術審評制度幾乎構成了各國藥品監管的核心。[3]然而我國藥品技術審評工作的整體狀況還不盡如人意,審評人員不足、審評資源匱乏等情況突出,且短期內難以徹底解決,審評時滯、申請積壓等現象嚴重,遭到社會各界的詬病。在當前藥品審評制度改革正在開展之際,通過構建科學完善的藥品審評質量管理規范(Good review practice, GRP)體系以整合資源、優化流程、提高藥品審評質量和效率,不失為一種有效的探索。美國作為世界上藥品監管水平較高的國家,其GRP體系經過近20年的發展已較為成熟。因此,本文詳細介紹美國GRP的相關情況以期對完善我國藥品審評質量管理規范提供借鑒。
1 美國藥品審評質量管理規范概述
1.1 政策背景
1992年,美國國會通過了《處方藥申報者付費法案》(The Prescription Drug User Fee Act,PDUFA),授權美國食品藥品監督管理局(Food and Drug Administration, FDA)向藥品申報者收取相應費用。FDA將相當部分的新增資源用于雇傭審評人員,僅在PDUFA I(PDUFA第一次獲得授權)期間,FDA新藥審評人員*這里的新藥審評人員指的是藥品審評和研究中心(CDER)、生物制品審評和研究中心(CBER)、監管事務辦公室(ORA)以及專員辦公室(OC)從事藥品審評工作的人員。從1992年的1 277人增加到1997年的1 990人。[4]之后,如何更好地保證審評質量、提高審評效率成為FDA面臨的新問題。此外,FDA針對當時藥品審評工作雖然出臺了諸多指南,但大多是對申請人提出各類建議或要求,缺乏對藥品審評機構和藥品審評人員的具體要求和規范。這使得FDA的審評工作程序不夠明確、透明,弱化了申請人在審評過程中的參與和監督作用。同時,審評報告也普遍存在內容、格式等方面的不統一、不規范現象,導致申請資料總結不準確、遺漏關鍵信息、可讀性差、結構化差異明顯等問題。[5]監管部門逐漸開始意識到這一問題,藥品審評和研究中心(Center for Drug Evaluation and Research, CDER)與生物制品審評和研究中心(Center for Biologics Evaluation and Research, CBER)于1996年共同提出了GRP的概念,旨在彌補這一監管的缺失。[6]
1.2 發展歷史
在GRP概念提出初期,其發展緩慢,最初幾年僅頒布了四項指南。[6]直至2005年4月,CDER和CBER根據PDUFA III(PDUFA第三次獲得授權)的目標要求制定了《審評人員和企業指導原則——PDUFA產品的藥品審評質量管理規范與實踐》(Guidance for Review Staff and Industry-Good Review Management Principles and Practices for PDUFA Products,GRPMs),該指導原則在后續工作中發揮了指導性作用。[7]之后,GRP進入快速發展期。2007年,《21世紀CDER審評程序參考指南》(The CDER 21st Century Review Process Desk Reference Guide,DRG)發布,提供了更多與GRMPs所規定的審評活動和時限相一致的信息,GRP體系更加趨于完善。[8]
目前,經過近二十年的發展,美國FDA已經頒布了總則、生物統計、臨床試驗、臨床藥理、藥理毒理、藥物安全性等多學科類別的指導性文件,基本形成了一個較為完整的GRP體系,成為美國藥品監督管理工作的重要組成部分。
1.3 現狀
藥品審評質量管理規范是基于FDA長期以來審評實踐的豐富經驗而制定的。指導性文件構成了這一體系的主體。這些指導性文件以“質量、效率、明確、透明、一致”為核心價值,規定了藥品審評過程中審評機構和審評人員的法定職責以及如何更加高效、科學地履行這些職責。[6]GRP體系的指導性文件涉及藥品和生物制品審評過程、審評管理和審評報告格式、內容等多個方面,覆蓋藥學、臨床、藥理、毒理等多個學科,貫穿申請提交前、受理及審評計劃制定階段、專家咨詢會議階段、審評階段、審評結論階段、審評結論后階段等各個審評階段,體系龐大。文件類型包括供企業和審評人員使用的指導原則,如上述的GRMPs等;CDER制定的政策及程序手冊(MAPP),如《藥品審評質量管理規范:統計審評模板》等;CBER制定的標準操作規程及政策手冊(SOPP),如《生物制品許可證(BLA)申請的管理流程》等。[9-10]這些指導性文件分別由CDER和CBER的相關辦公室編寫,提供了有關CDER和CBER的具體工作流程、審評人員工作要求以及給企業的建議等細節。
2 美國藥品審評質量管理規范的政策目標和相應措施
2.1 政策目標
在GRP概念提出前,早期的FDA藥品審評過程中藥品審評人員的工作職責、工作程序以及審評活動中積累的知識和經驗并未明確地以文字形式記錄下來。[6]藥品審評質量管理規范正是致力于將審評過程中積累的現有知識、經驗進行系統化、書面化,對審評過程進行規范化,以到達雙重目標[7]:即一方面保證藥品審評質量,另一方面提高藥品審評效率。
2.2 相應措施
為達成上述目標,GRP提供了諸多措施,包括明確了在做出監管決策之前必須完成的關鍵審評步驟和相應時限,出臺指導原則和審評模板以保證審評過程、審評關注點以及審評報告格式的一致性,重視培訓和績效評估以推動GRP的貫徹實施和持續改進。
這些措施共同彰顯了GRP的核心價值——質量、效率、明確、透明和一致,既保證了審評過程和審評管理的質量和一致,又提高了審評過程和審評管理的效率、清晰度和透明度。[11]
2.2.1 明確審評過程和相應時限
時限制度通過對藥品審評過程中的審評人員和申請人的行為給予時間上的限制,可以有效保障審評效率和申請人合法權益。[12]FDA估計,審評完成每提前1個月,生產商的平均成本節約1千萬美元。[13]GRMPs系統梳理了整個藥品審評過程,將藥品審評過程分為受理和審評計劃、審評、咨詢委員會會議、審評結論和審評結論后五個階段,將涉及到的審評活動細分為41個關鍵事項,明確規定了相應的完成時限,并指定了負責部門和個人。
以受理和審評計劃階段為例(表1),GRMPs規定了文件管理部門、項目經理以及審評小組成員需要完成的關鍵事項和相應時限,如應在接收申請材料后的14天內,指定項目經理,開始立卷審查,發出申請受理書面通知,指定審評小組。明確細致的工作內容、審評時限以及責任主體為審評人員提供了行動指南,也使申請人及時掌握審評動態、實時監督審評進程,進而提高藥品審評工作的質量和效率。
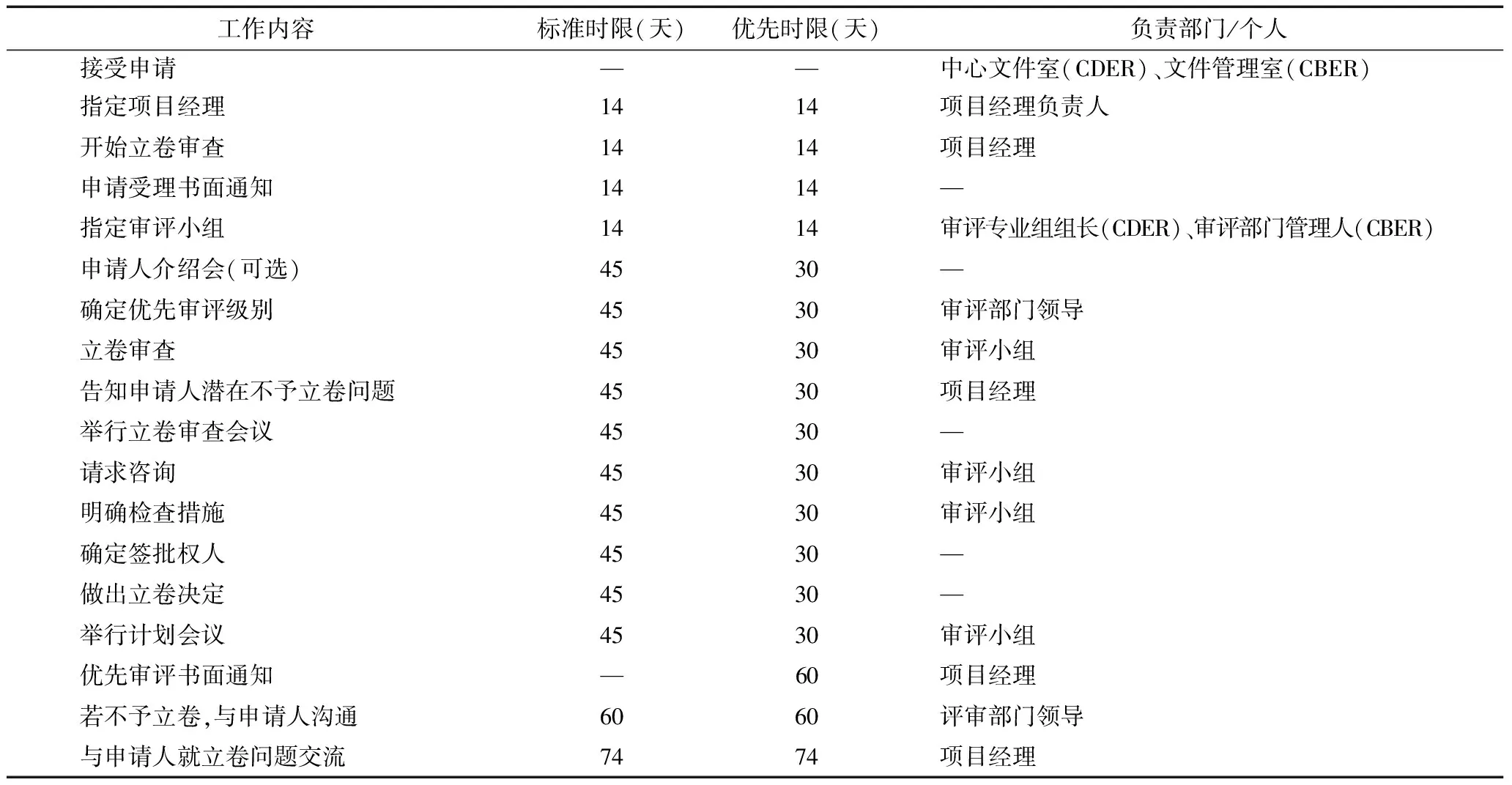
表1 受理及審評計劃階段的關鍵事項及相應時限[7,14]
2.2.2 出臺各專業審評模板
藥品審評工作作為一項高度智力化的活動,具有很強的靈活性和主觀性,然而審評結果的一致性和客觀性又是我們追求的目標。面對這一矛盾,GRP提供了各專業審評的模版,并細分為不同申報情況下的撰寫要求,分別指導相應學科的專業審評工作。審評模板盡可能將審評過程中的共性部分實行嚴格控制,以保證審評過程的一致性。
這些審評模板明確了審評過程中應關注哪些資料,如何將相關技術考慮體現在審評報告中,如《藥品審評質量管理規范:統計審查模板》用于記錄生物統計學室(OB)對新藥上市申請(New drug applications, NDAs)和生物制品許可申請(Biological License Applications, BLAs)的初步統計審評結論,規定審評報告應包括執行概要、引言、統計評價、特殊人群/亞組人群的發現、總結和結論等部分,并詳細說明了各部分所占篇幅、撰寫規范和要求等,細致且可操作性較強。[15]
一般情況下,這些審評模板在各專業審評過程中是強制執行的,為了適應特殊事項或特定程序,各臨床審評部門可對模板進行修改。然而,這些修改必須在整個部門做到標準化、一致化,在模板更改控制委員會備案,并且將其作為原文件的修訂版執行。[16]
2.2.3 貫徹實施和持續改進
任何改革政策的推行都不是一蹴而就的,僅制定一套藥品審評質量管理規范無法立即帶來正面的變化,還需要不斷的相關培訓和績效評估改進,方能貫徹實施和持續改進。[7]
FDA的培訓包括專業素質培訓和共性能力培養,前者包括藥學、臨床、藥理毒理、法律法規等專業知識培訓;后者包括分析比較能力、審慎思考能力、溝通交流能力、處理分歧能力等培養。[5]另外FDA還經常性地組織各類內外部交流活動,討論學習、共享經驗,并形成記錄資料供工作人員隨時學習。
為評估GRP的實施情況并進行持續改進,GRP規定應定期進行績效評估。如2011年,FDA曾委托博思艾倫咨詢公司(Booz Allen Hamilton)通過審閱審評記錄考察61項NDAs和BLAs的審評過程中審評人員對GRMPs所規定的41項關鍵活動和相應時限的遵從情況。同時結合與監管項目經理、一線審評人員和申請人代表的訪談、FDA工作人員問卷調查,分析可能影響GRP貫徹實施的因素,并給出相應的改善建議。[14]
3 美國藥品審評質量管理規范的實施效果
美國GRP經過近二十年的發展,其對藥品審評工作的影響日益凸顯,基本實現了政策初衷,在保證藥品審評工作質量,確保擬批準上市藥品安全、有效、質量可控的前提下,大幅縮短了藥品審評時間、顯著提高了首輪藥品批準率。
3.1 大幅縮短藥品審評時間
藥品審評時間作為藥品審評工作的一項重要指標,可以反映藥品審評工作的成效,也是GRP實施效果的直觀體現。[17]從圖1可以看出,從1996年起,除少數年份外,美國CDER和CBER對NDAs和BLAs的審評時間整體大幅縮短:1993年標準審評時間中位數為28個月,優先審評時間中位數為13.2個月,1996年GRP施行后,標準審評時間中位數下降至12.1個月,優先審評時間中位數降至6個月,2013年標準審評時間中位數降至10個月、優先審評時間中位數保持在6個月。
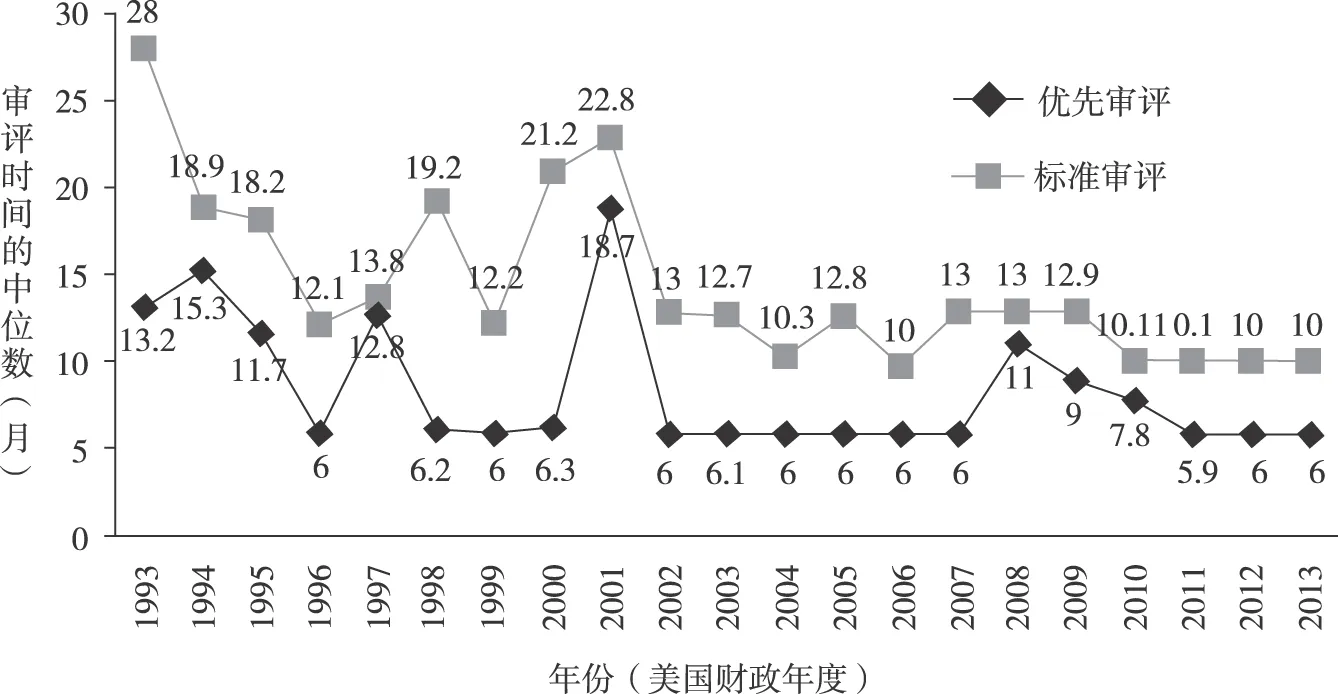
圖1 1993—2013年CDER和CBER的NDAs和BLAs審評時間中位數[18-19]
3.2 顯著提高首輪藥品批準率
如圖2 所示, CDER和CBER的NDAs和BLAs的首輪藥品批準率從1993年的36%升至1996年的50%,近幾年穩定保持在70%~75%之間。博思艾倫咨詢公司通過對185項新藥申請(這些申請均是于2002—2007年接收且在2007年9月30日前做出審評結論,其中NDAs占74%,BLAs占26%)的審評過程和審評結論進行相關性分析發現,首輪藥品批準率與審評過程中審評人員對GRMPs所規定的關鍵事項及相應時限的遵從率*遵從率指一項新藥審評過程中,審評人員在規定時限內完成的關鍵事項數與GRMPs規定的關鍵事項總數(41項)的比值。正相關:遵從率為80%的新藥申請,其首輪藥品批準率為71%,而遵從率為20%的新藥申請的這一比例僅為50%。[20]由此可見,明確并遵守藥品審評過程的關鍵事項和相應時限,有利于提高首輪藥品批準率,減少藥品審評周期數,從而提高藥品審評效率。
4 啟示與建議
4.1 我國藥品審評質量管理規范的現狀
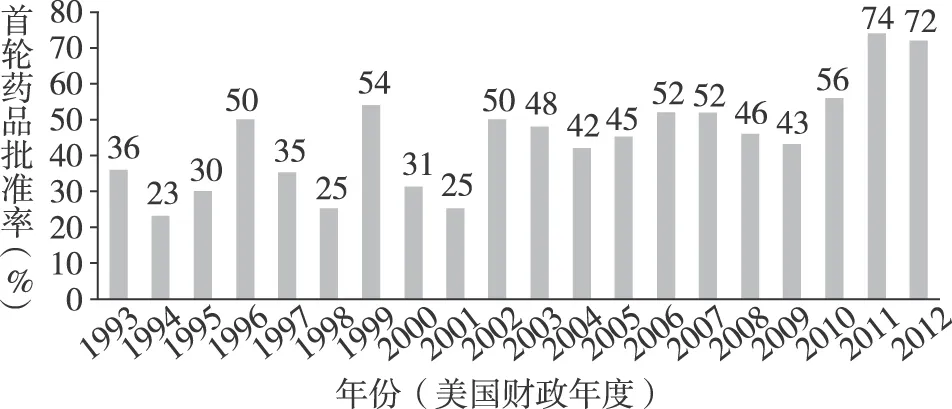
圖2 1993—2013年CDER和CBER的NDAs和BLAs首輪藥品批準率[18]
近年來,國家食品藥品監督管理總局藥品審評中心根據《行政許可法》、《藥品注冊管理辦法》等法律法規的要求,結合藥品審評工作實際,出臺了諸多規范。2011年出臺的《藥品技術審評原則和程序》,為藥品審評工作和制度規范化建設提供了依據,并就構建我國藥品審評質量管理規范體系提出了要求。[21]隨后,藥品審評中心先后制定并出臺了《藥品審評中心審評任務管理規范(試行)》和《藥品審評中心技術審評決策路徑管理規范(試行)》等一系列規范性文件。[22]較之以往,我國藥品審評工作更加公開、透明、科學、民主。但是目前我國藥品審評質量管理規范體系尚未完全建立,還存在諸多不足。主要表現在以下幾個方面:
第一,我國只籠統地規定了藥品審評時限,一方面這些時限規定并不符合現實情況,另一方面缺乏詳細的審評步驟和相應時限規定,導致這些籠統的時限規定難以落實,形同虛設,“審評超時”問題一直未能得到有效改善。使申請人不能清晰把握所申報品種的審評節奏,廣大群眾更是缺乏監督的具體量化標準。[23]
第二,我國藥品審評中心雖然出臺過審評報告撰寫意見,但未體現科學、統一的審評原則和尺度,也未將審評過程中積累的經驗標準化、制度化,導致審評報告完整性差、結構性差異明顯,甚至因信息遺漏而導致出現審評質量問題。
第三,目前,我國藥品審評部門的培訓制度大多為短期、零散且不成體系。新的法律、法規出臺后,也多以員工自我學習為主,這種方式不利于新政策的貫徹施行。
第四,我國藥品審評質量管理規范沒有相應評價體系,也未能及時更新,個別制度規范已不符合現實情況,某些新情況、新問題又無章可循,導致審評工作出現不同程度的管理風險,難以保證審評工作的科學性、規范性和一致性。
4.2 完善我國藥品審評質量管理規范的建議
4.2.1 細化我國藥品審評時限規定
審評時限是藥品技術審評工作的重要內容,也是申請人和社會各界非常關注的問題。我國2007版《藥品注冊管理辦法》第一百五十條規定新藥臨床試驗申請和新藥上市申請的標準審評時限分別為90日和150日,而《2013年度藥品審評報告》統計數據顯示,兩者的實際審評時間分別為9個月和19個月*新藥臨床試驗和新藥上市申請的實際審評時間是按藥品審評中心系統顯示的“等待時間”加上“法定審評時間”(以30天/每月計算)得到。,巨大的差距顯示出我國藥品審評時限規定的不科學、不合理。因此,應進一步完善我國藥品審評時限規定,結合我國藥品注冊申請基數大、積壓嚴重、審評資源匱乏的實際情況,修改相關法律法規中的時限規定,使之更加切實可行、科學合理。在此基礎上借鑒美國GRMPs中的藥品審評關鍵步驟和相應時限規定,結合我國藥品審評工作實際,梳理審評流程、識別關鍵節點,細化時限規定。
4.2.2 制定可操作性強的審評模板
藥品審評作為技術性、專業性、公共性服務行為,應體現科學、公平和一致,因此,制定規范化、可操作性強的審評模板至關重要。建議首先由各專業資深審評人員總結其在審評過程中積累的經驗,明確審評關注點、審評原則和尺度以及審評報告應包括的內容等。然后按藥品種類、申請類別、適應癥等提煉并標準化這些最佳操作規范,制定或修訂審評模板,使之反映藥品審評規律且利于操作。在審評過程中,審評人員可就審評模板的適宜性進行討論,也可聽取申請人的建議,在實際操作中不斷完善審評模板,使其更加科學、規范、可操作。
4.2.3 重視藥品審評質量管理規范體系的相關培訓和持續改進
審評人員培訓是藥品審評質量管理規范貫徹實施的重要途徑,建議借鑒美國的培訓經驗構建系統、高效、統一、經濟的藥品審評人員培訓模式。具體而言,培訓內容應包括專業知識培訓、共性能力培訓、審評規范培訓以及職業道德培訓等。同時應加強培訓工作的統一性,尤其是對法律法規、審評規范的培訓。另外,可采用多元化培訓形式,例如網絡培訓、“導師制”新審評人員培訓等。
規范性文件的制定大多立足于當時的現實情況,隨著社會政治經濟條件的變化,必然會出現不再適用的部分。主管部門應盡快出臺“制度文件的立、改、廢工作指南”,定期對藥品審評質量管理規范體系中的指導性文件進行適用性評估和修訂。另外,應借鑒美國經驗構建獨立的追蹤評價體系,及時發現制度實施中的問題,尋求持續改進。
[1] 韋冠. 國內外藥品上市許可制度比較及借鑒[J]. 中國藥房, 2008, 19(34): 2650-2653.
[2] 武志昂, 畢開順. 從市場經濟角度看藥品注冊管理[J]. 中國藥師, 2007, 10(5): 489-491.
[3] Jason P, Peter D, Ian W. Medico legal Essentials in Heal-thcare[M]. Churchill Livingstone, 2004.
[4] Office of Management and Systems of FDA. PDUFA III Five-Year Plan [EB/OL]. (2003-07-27) [2014-11-15]. http://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM176324.pdf
[5] 呂東, 陳曉媛, 黃文龍. 中美藥品技術評價體系比較[J]. 中國新藥雜志, 2009, 18(2): 98-104.
[6] 宋華琳. 美國藥品審評質量管理規范評介[J]. 藥學進展, 1999, 23(6): 350-353.
[7] CDER of FDA, CBER of FDA. Guidance for Review Staff and Industry:Good Review Management Principles and Practices for PDUFA Products [EB/OL]. (2005-03-15) [2014-11-15]. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm079748.pdf
[8] CDER of FDA. The CDER 21st Century Review Process Desk Reference Guide (DRG) [EB/OL]. (2014-09-10) [2014-11-15]. http://www.fda.gov/downloads/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/manualofpoliciesprocedures/ucm218757.pdf
[9] CDER of FDA. Good review practice [EB/OL]. (2013-12-02) [2014-11-15]. http://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/ucm118777.htm
[10] CDER of FDA. Biologics Procedures (SOPPs) [EB/OL]. (2010-05-19) [2014-11-15]. http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/ProceduresSOPPs/default.htm.
[11] Office of new drugs of FDA. Good review practices (MAPP 6025.1) [EB/OL]. (2012-03-02) [2014-11-15].http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalPr o ductsandTobacco/CDER/ManualofPoliciesProcedures/ucm082016.pdf
[12] 宋華琳. 中國藥品審評法律制度的行政法改革[J]. 行政法學研究, 2014(3): 3-12.
[13] 姚立新, 李茂忠, 董江萍, 等. 從PDUFA I到PDUFA V——FDA通過法規體系的完善實現新藥審評的持續改進[J]. 中國新藥雜志, 2013, 22(10): 1143-1156.
[14] Booz Allen Hamilton.Assessment of GRMPs Implementation [EB/OL]. (2011-04-29) [2014-11-15].http://www.fda.gov/downloads/forindustry/userfees/prescriptiondruguserfee/ucm272446.pdf
[15] Office of Translational Sciences of FDA. Good Review Practice: Statistical Review Template (MAPP 6010.4)[EB/OL]. (2012-07-30) [2014-11-15]. http://www. fda.gov/downloads/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/manualofpolicies- procedures/ucm313814.pdf
[16] Office of New Drug of FDA. Good Review Practice: Clinical Review Template (MAPP 6010.3) [EB/OL]. (2010-12-10) [2014-11-15]. http://www.fda.gov/downloads/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/manualofpoliciesprocedures/ucm080121.pdf
[17] 董江萍, 劉璐, 張象麟. 中美兩國藥品注冊工作時限管理的比較[J]. 中國新藥雜志, 2005, 14(1): 4-7.
[18] Jenkins J K. CDER New Drug Review:2012 Update [EB/OL]. (2012-12-10) [2014-11-29]. http://www.fda. gov/downloads/About%20FDA/Centers%20Offices/Office%20of%20Medical%20Products%20and%20Tobacco/CDER/UCM331454.pdf
[19] FDA. FY 2013 Performance report to the president and congress for the Prescription Drug user Fee Act [EB/OL]. (2014-04-23) [2014-11-29]. http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/UCM384035.pdf
[20] Booz Allen Hamilton. Independent Evaluation of FDA’s First Cycle Review Performance-Final Report [EB/OL]. (2008-07-16) [2014-11-15]. http://www.fda. gov/downloads/ForIndustry/UserFees/PrescriptionDrugUs-erFee/ucm127982.pdf
[21] 國家食品藥品監督管理總局藥品審評中心. 藥品技術審評原則和程序[EB/OL]. (2011-03-23) [2014-11-30]. http://www.cde.org.cn/news.do?method=large- Info&id=312103
[22] 國家食品藥品監督管理總局藥品審評中心. 關于印發《藥品審評中心技術審評決策路徑管理規范(試行)》和《藥品審評中心審評任務管理規范(試行)》的通知【藥審業[2011]129號】[EB/OL]. (2011-10-09) [2014-11-30]. http://www.cde.org.cn/regulat.do?method=largePage&id=2851
[23] 李艷坤, 邢花, 陳宇, 等. 芻議我國藥品技術審評原則和程序的新變化[J]. 中國新藥雜志, 2012, 21(10): 1073-1075.
(編輯 趙曉娟)
Evaluation on good review practice in the United States of America and its implications for China
GENGXiao-ya,SHAORong
InternationalPharmaceuticalBusinessSchool,ChinaPharmaceuticalUniversity,NanjingJiangsu211198,China
The paper summarizes the background, development history and current state of good review practice (GRP) in the United States of America. On this basis, the paper then focuses on measures for the realization of GRP policy objective and introduces the effects of GRP by using the statistical data of the median time to application approval and approval rates of New drug applications(NDAs) and Biological License Applications(BLAs) on the first cycle. Through research, the paper considers a set of scientific and comprehensive GRP that can effectively guarantee the quality of drug reviews and improve the efficiency of drug reviews. However, China’s GRP is not complete yet. So it also suggests that improving GRP in China further by detailing the timeline of drug review in China, developing workable review templates, emphasizing training and the continuous improvement of GRP.
The United States of America; Drug evaluation; Good review practice
耿曉雅,女(1991年—),碩士研究生,主要研究方向為醫藥政策與法規研究。E-mail:gengxiaoya52@163.com
邵蓉。E-mail:shaorong118@163.com
R197
A
10.3969/j.issn.1674-2982.2015.02.010
2014-11-24
2014-12-21
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
北部灣大學學報(2022年1期)2022-06-22 04:58:38
北部灣大學學報(2022年2期)2022-06-21 11:44:36
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
現代儀器與醫療(2021年4期)2021-11-05 08:25:08
北部灣大學學報(2021年4期)2021-04-28 08:01:04
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
中國衛生(2016年5期)2016-11-12 13:25:28
汽車觀察(2016年3期)2016-02-28 13:16:26
