在堿金屬/液氨作用下兩類還原反應的機理探討
2015-02-13 09:27:36戴亞中裴堅
大學化學 2015年4期
戴亞中 裴堅
(北京大學化學與分子工程學院 北京100871)
除金屬鈹以外的堿金屬和堿土金屬均可溶解在液氨中并形成一種藍色溶液。堿土金屬和堿金屬溶解于液氨中均會生成氨合金屬正離子(M(N)和氨合電子(e(N)。這種處在亞穩狀態的溶液具有很強的還原性(e(NH3的標準電極電勢為-2.86V)。但是,這種溶液會逐漸分解,產生金屬氨基化物和氫氣。在低溫條件下,分解反應的速率比較緩慢;但在有過渡金屬鹽催化時,分解反應會加速進行。這種具有強還原性的堿金屬/液氨溶液在有機合成中得到廣泛的運用,主要包括兩類還原反應:炔烴被還原成烯烴的反應和Birch還原反應,一般認為它們在反應過程中都生成了自由基負離子中間體。我們將在本文中對它們的機理進行進一步探討。
1 堿金屬/液氨下炔烴的還原反應
炔烴用堿金屬和液氨還原生成烯烴的反應,是有機化學中合成E型烯烴衍生物的經典方法之一。例如在圖1中,2-丁炔可以通過此還原反應生成E型2-丁烯[1]:

圖1 2-丁炔用鈉和液氨還原生成E型2-丁烯
反應的過程如圖2所示:首先是炔烴分子(1)中的碳碳三鍵從液氨中得到一個氨合溶劑化電子生成烯基負離子自由基(2)。烯基負離子自由基(2)是一種很強的堿,能夠從液氨中奪取一個質子生成E型烯基自由基(3),該烯基自由基繼續從堿金屬處得到一個電子生成烯基碳負離子(4)后,4再從液氨中奪取一個質子生成最終產物E型烯烴(5)[1]。
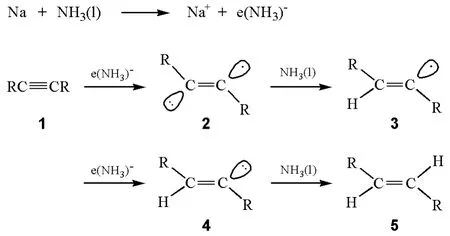
圖2 炔烴用鈉和液氨還原的機理
產物烯烴的立體構型是在化合物1轉化為3的過程中建立的。在反應過程中,首先生成了烯基自由基負離子,然后2得到質子后生成烯基自由基(3),接著生成空阻較小的反式烯基負離子(4),因此反應產物的構型為E型。通過ESR(電子自旋共振譜)實驗的測量發現烯自由基的E/Z構型的翻轉能壘很低,約為12kJ/mol[2],因此烯基自由基可以進行快速的順式和反式之間的平衡,但是烯基負離子的順式和反式的轉換要慢得多。因此,更為穩定的反式烯基負離子就比順式的烯基負離子更容易生成,它在形成平衡前就被質子化生成E型烯烴。該反應條件中溶劑為液氨,是一種強極性的溶劑,對反應中間體(2和3)有強的溶劑化作用,可以進一步穩定反應中間體,使這些中間體的構型不容易改變。
此外,由于烷基自由基負離子較乙烯基自由基負離子穩定性差,因此,得到的烯烴難于在此條件下繼續被還原成烷烴。末端炔烴不適用于此條件下的還原,因為末端炔烴的質子有著強的酸性,在此條件下優先發生酸堿中和反應,生成炔烴負離子,由于負電荷之間的互相排斥作用,炔烴負離子不易繼續從溶液中得到電子而被還原。
綜上所述,炔烴在堿金屬和液氨還原條件下的反應機理主要是從中間體(2)開始,其新形成的雙鍵構型主要為反式,最終決定了產物為E型烯烴。烯烴在此條件下不會繼續被還原至烷烴。
2 Birch還原反應
Birch還原反應是有機化學中一個重要的人名反應,一般認為是指芳香族化合物在液氨和醇的混合液中被堿金屬(鈉、鋰或鉀)通過1,4-還原至非共軛的1,4-環己二烯的反應(圖3)。雜環化合物(如吡啶、吡咯或呋喃類衍生物)也可以被還原。使用的醇通常是乙醇或叔丁醇。醇此時作為質子供體,為反應提供氫的來源。還原產物1,4-環己二烯中的孤立碳碳雙鍵在這個條件下不會繼續被還原至飽和烷烴。此外,Birch還原反應也有一些缺陷:富電子體系的雜環芳烴需要至少有一個吸電子取代基存在才能被還原。例如呋喃、噻吩不能被還原,只有其環上有吸電子取代基存在的條件下時才能被還原。

圖3 Birch還原
在《基礎有機化學》(第3版)中,對Birch反應做出了如下說明:“給電子基團一般導致還原速率減慢;吸電子基團使得還原速率加快,而且所得產物取代基團位于被還原的碳原子上”[1],下面對它的還原位置的選擇性和反應的決速步進行探討和理清。
研究表明,與炔烴被還原時一樣,由于堿金屬溶解于液氨,所得的溶液含有強還原性的溶劑化電子。底物中的苯環可以接受電子,形成的自由基負離子接著從溶劑中得到質子,不可逆地形成環己二烯自由基,環己二烯自由基再接受一個電子生成環己二烯負離子,最后環己二烯負離子的質子化過程發生在中心碳原子上,形成非共軛雙烯——1,4-環己二烯。Birch A J等人通過計算發現己二烯負離子中間體HOMO的最大軌道系數在中間碳原子上,氫離子優先與位于中間的碳原子結合,生成1,4-環己雙烯。因此Birch還原往往得到熱力學不穩定的非共軛1,4-加成產物。而不是熱力學穩定的1,3-加成產物[3]。此外,依據芳香族底物上取代基的位置,電子加成的位置也會有所不同。因此,Birch還原可能的機理有鄰位進攻(ORTHO)、間位進攻(META)以及對位進攻(PARA)3種[4](圖4)。
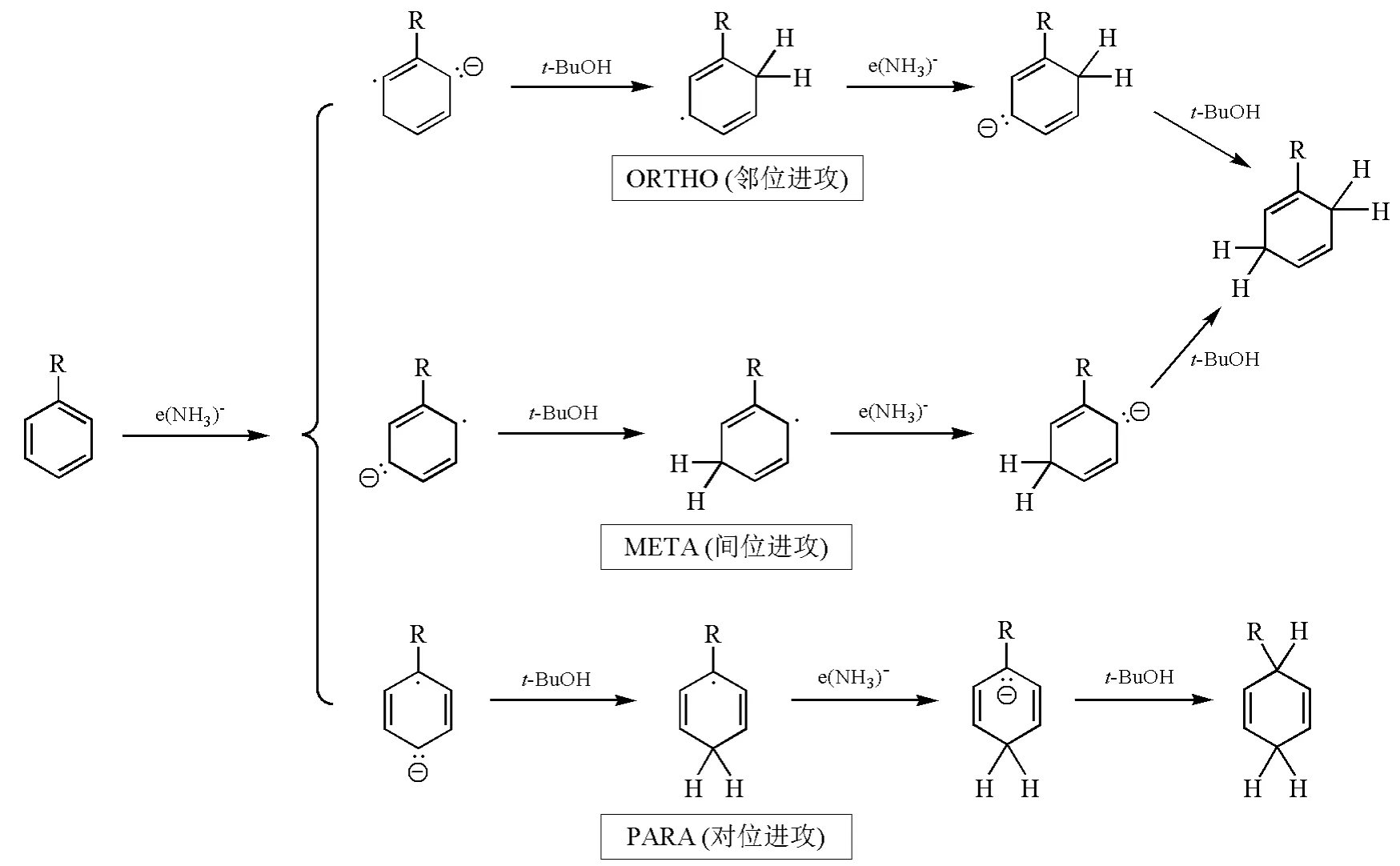
圖4 Birch還原的可能途徑示意圖
總的來說,芳香化合物中的苯環通過Birch還原生成1,4-環己二烯的反應選擇性主要取決于苯環上取代基的性質。當取代基為吸電子基時,往往采取PARA途徑,此時反應在能量上更為有利,中間體能通過共振方式穩定碳負離子;當取代基是供電子基時,采取ORTHO或者META途徑對于反應的進行更為有利。如圖5所示,Zimmerman H E課題組以甲基苯和甲氧基苯為例,通過計算,比較了反應中間體自由基的能量,發現盡管ORTHO和META兩種反應方式的最終產物相同,但是計算結果表明ORTHO途徑的自由基中間體更為穩定[5]。Zimmerman H E課題組在還原甲氧基苯時,利用氘標記的特丁醇提供質子,通過測定反應產物氫原子同位素的位置及比例,以實驗證明了甲氧基苯等富電子體系還原時以ORTHO方式為主[5]。
從Birch還原反應結果上看,相當于電子和R+(R可以是氫以外的物種)對苯環進行了1,4-加成(圖6)。吸電子基團有利于電子對苯環的加成,因此能夠加速反應的進行;給電子取代基則相反。當苯環上同時有給電子基團和吸電子基團的時候,Birch還原的選擇性主要取決于吸電子基團[5]。
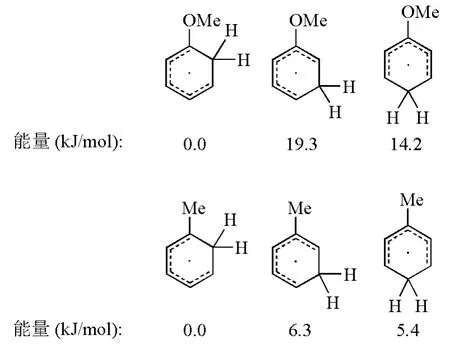
圖5 自由基中間體的能量比較計算基組為B3LYP/6-3G*
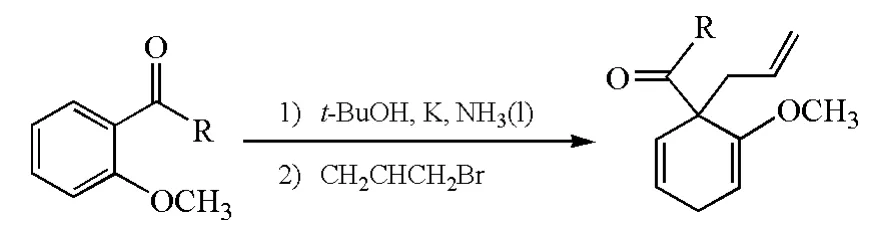
圖6 多取代苯進行Birch還原時的反應選擇性
Krapcho小組進行了測定Birch還原反應的反應速率常數的一系列實驗,發現Birch還原反應在化學動力學上為三級反應,反應速度正比于芳香底物的濃度、溶劑化電子的濃度以及作為氫源的醇的濃度。通過動力學擬合,Krapcho等人認為圖3中第二步,即環己二烯負離子與醇生成環己二烯自由基的反應為Birch還原反應的決速步。芳香體系上的吸電子取代基會有利于環己二烯負離子的生成,因此能提高Birch還原的反應速率。對于富電子的吡咯、噻吩等芳香化合物以及給電子基團取代的苯衍生物,如果芳環上沒有吸電子取代基的話,環己二烯負離子不易生成,此時Birch還原反應速度很慢[6]。
考慮到Birch還原會生成一個負離子中間體,可以被一個合適的親電試劑捕獲,科學家在Birch還原的基礎上,發展了Birch烷基化反應:在鹵代烴的存在下,在Birch還原過程中得到的碳負離子可作為親核試劑,經親核取代反應生成新的碳碳鍵(圖7)。

圖7 Birch烷基化反應
Birch還原從反應過程中可以理解為芳環的1,4-還原,其產物為非共軛的1,4-環己二烯,孤立碳碳雙鍵不會被還原成飽和烷烴。這是因為乙烯的最低未占有軌道(LUMO)能級為-1.5eV,e(NH3)x-的標準電極電勢為-2.86 V,生成的乙烷負離子能量很高,不穩定,因此非共軛的雙鍵不會被液氨中的溶劑化電子還原[7-8]。如果非芳香性的有機化合物LUMO能級足夠低的話,它也可以在Birch還原的條件下被還原。Wooster C B與Godfrey K L等人發現對于共軛烯烴、α,β-不飽和羰基化合物、雙取代炔烴、共軛炔烴和苯乙烯等體系,由于有共軛體系能穩定負電荷,它們也能夠在液氨中被堿金屬還原(圖8)[9],這些反應已在有機合成中得到廣泛的運用和推廣[10]。
在圖8中的α,β-不飽和羰基化合物的烷基化反應過程中,羥基與氨分子會形成氫鍵,導致氨上質子定向從羥基方位附近被奪取;在后續的烷基化過程中,烯醇負離子從位阻小的方位進攻鹵代烷,反應有很高的立體選擇性(圖9)。

圖8 Birch還原實例
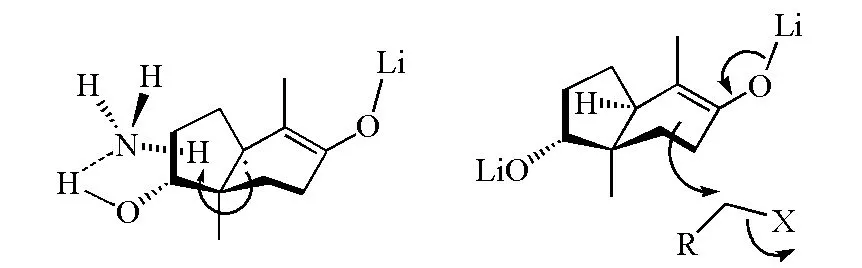
圖9 Birch還原實例(圖8(a))的反應構象
3 總結
堿金屬還原反應在有機化學中應用廣泛,本文結合該反應在《基礎有機化學》[1]上的部分涉及內容(炔烴還原和Birch還原),調研文獻,對它的反應機理進行了討論,總結了這些反應生成特定產物的原因。
[1]邢其毅,裴偉偉,徐瑞秋,等.基礎有機化學.第3版.北京:高等教育出版社,2009
[2]Fessenden R W,Schuler R H.J Chem Phys,1963,39(9):2147
[3]Birch A J,Hinde A L,Radom L.J Am Chem Soc,1980,102:6430
[4]Birch A J.J Am Chem Soc,1944,430
[5]Zimmerman H E.Acc Chem Res,2012,45:164
[6]Krapcho A P,Bothner-By A.J Am Chem Soc,1959,81:3658
[7]Lagowski J J.現代無機化學(上冊).孟祥勝,許丙安譯.北京:高等教育出版社,1984
[8]李彥如.承德民族師專學報,2004,24(2):42
[9]Wooster C B,Godfrey K L.J Am Chem Soc,1937,59(3):596
[10]Kaplan H Z,Rendina V L,Kingsbury J S.J Org Chem,2013,78:4620
