低溫固相法制備的納米α-MnO2的性能
2015-01-16 08:39:14吳昊天張振忠趙芳霞王林燕
電池 2015年3期
吳昊天,張振忠,趙芳霞,王林燕
(南京工業大學材料科學與工程學院,江蘇南京 210009)
與鋰離子電池相比,二次鋅離子電池的充電時間短、衰減慢,且價格低廉,無毒安全,同時也具有高比容量。鋅離子電池用的α-MnO2屬于一維隧道T[2×2]結構,能容納較大的離子(如K+)[1],Zn2+在隧道中的嵌脫較容易。目前,已有多種方法用于制備α-MnO2。張偉等[2]以高錳酸鉀和乙酸錳為原料,用機械化學法制備弱結晶型的塊狀和球狀間雜的α-MnO2,粒徑為2 ~10 μm;薛兆輝等[3]以高錳酸鉀、氧化石墨和硫酸為原料,用水熱法合成α-MnO2納米棒,直徑為10~20 nm、長度為300~400 nm;汪形艷等[4]以氧化鋁膜為模板,用溶膠-凝膠法制備直徑約為70 nm,長度為500~700 nm的納米線。α-MnO2具有良好的電容性能,但用于電池的研究很少,原因是α-MnO2不導電,用作電極材料,需要較多的導電劑;其次,MnO2的晶型復雜多樣。
本文作者以高錳酸鉀(KMnO4)和四水合氯化錳(MnCl2·4H2O)為原料,采用低溫固相法制備鋅離子電池正極材料納米α-MnO2,測試制備的鋅離子電池的電化學性能。
1 實驗
1.1 α-MnO2的制備及分析
將 KMnO4(江蘇產,AR)和 MnCl2·4H2O(廣東產,AR)在瑪瑙碾缽中各自研細,再按物質的量比為2∶2、2∶3和2∶4混合,研磨約30 min,當混合物由紫色轉變為棕褐色或黑色時,轉移到DC-2006低溫恒溫槽(上海產)中,在80℃下水浴中加熱3 h,之后,用去離子水和無水乙醇(江蘇產,AR)離心洗滌數次,去掉沉淀,在80℃下烘干至恒重,得到的產物分別記為樣品S-1、S-2和S-3。
用GENESIS 2000 XMS型X射線衍射儀(美國產)和Tecnai G2 20 S-TWIN透射電子顯微鏡(TEM,美國產)分別檢測制備粉體的相結構、顆粒形貌與團聚狀況。
1.2 電池的組裝及性能測試
將質量比 7∶2∶1的 α-MnO2、乙炔黑(國藥集團,CP)和聚四氟乙烯(江蘇產,CP)加水攪拌后,涂覆于泡沫鎳(江蘇產,電池級,110PPI)上,用769YP-15A型粉末壓片機(天津產)以16 MPa的壓力壓制1 min,在60℃下烘干至恒重,制得尺寸為2.0 cm×2.0 cm×0.6 cm的正極(約含0.09 g活性物質),在端點處焊接一條鎳帶。負極以超細鋅粉(江蘇產,AR)為活性物質,用相同的方法制作(約含0.1 g活性物質)。
將正、負極插入1 mol/L ZnSO4(上海產,AR)溶液中,以聚丙烯膜(上海產,電池級)為隔膜,組裝半徑2 cm、高4 cm的模擬電池。由樣品S-1、S-2和S-3組裝的模擬電池,分別編號為cell-1、cell-2和cell-3。
以α-MnO2電極為研究電極、鋅電極為對電極和參比電極,用CHI660A電化學工作站(上海產)進行循環伏安測試,1號電池的電位為0.8~2.3 V,2、3號電池的電位為1.0~2.3 V,掃描速率均為0.5 mV/s。用2001A電池測試系統(成都產)進行恒流充放電測試,首先測試不同電流(50 mA/g、100 mA/g、200 mA/g、500 mA/g 和 1 000 mA/g)時電池的首次放電比容量,放電至電壓為0.3 V;再測試電流為100 mA/g時電池10次循環的放電比容量,電壓為2.0~1.0 V,充電電流均為100 mA/g。
2 結果與討論
2.1 結構與形貌
圖1為制備的納米α-MnO2樣品的TEM圖。

圖1 制備的納米α-MnO2樣品的TEM圖Fig.1 Transmission electron microscope(TEM)photographs of prepared nano α-MnO2samples
從圖1可知,各樣品均為納米棒狀顆粒。樣品S-1的顆粒尺寸最小,直徑約為7 nm、長度約為50 nm,長徑比約為7;樣品S-2的直徑約為9 nm、長度約為80 nm,長徑比約為9;樣品S-3的直徑約為10 nm、長度增加到120 nm左右,長徑比為12。隨著原料中KMnO4與MnCl2·4H2O物質的量比的提高,納米棒的直徑和長徑比均減小,尤其是縱向尺寸。
圖2為制備的納米α-MnO2樣品的XRD圖。
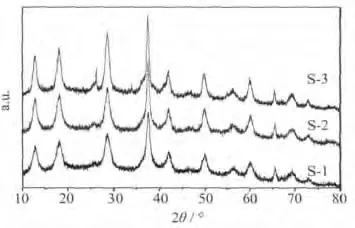
圖2 制備的納米α-MnO2樣品的XRD圖Fig.2 XRD patterns of prepared nano α-MnO2samples
從圖2可知,各樣品的XRD圖相似,在12.8°、18.1°、28.8 °、37.5 °、41.9 °、49.8 °、60.2 °和 68.2 °等位置出現了尖銳的特征峰。對比α-MnO2標準卡(PDF:44-0141)可知,樣品幾乎具有所有α-MnO2晶體的特征峰,未出現其他錳的氧化物雜質峰,說明均為高純度的α-MnO2晶體。樣品S-1、S-2和S-3在26.1°處的衍射峰逐漸銳化,在28.8°、37.5°等處的特征峰強度也逐漸增大,表明隨著原料中KMnO4與MnCl2·4H2O物質的量比的提高,產物的結晶性能提高。
2.2 電池性能測試
組裝的模擬電池的循環伏安曲線見圖3。
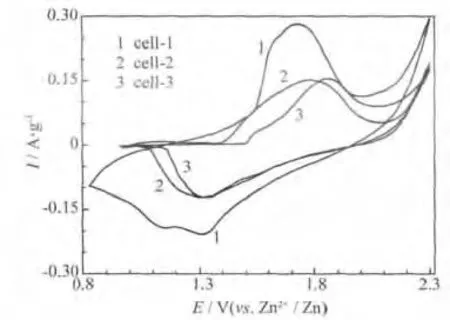
圖3 組裝的模擬電池的循環伏安曲線Fig.3 CV curves of assembled simulating cells
圖3中,曲線在1.2 V附近出現的還原峰,對應著放電過程中Zn2+嵌入α-MnO2隧道的行為;在1.7 V附近出現的氧化峰,對應著充電過程中Zn2+脫出MnO2隧道的行為。這種嵌脫的過程可用式(1)來描述。
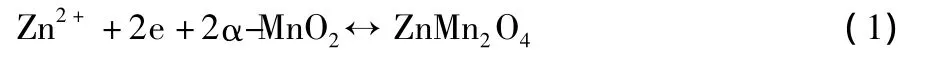
各電池的氧化還原峰電位差略有差別,其中,cell-1在1.714 V出現了寬化的氧化峰,在1.320 V處出現對應的還原峰,氧化還原峰電位差為0.396 V;cell-2、cell-3的氧化還原峰電位差分別為0.442 V、0.528 V。相比而言,cell-1的峰電流較高。較大的電位差,反應了較嚴重的極化現象,也代表著氧化還原可逆性不理想,因此,KMnO4與MnCl2·4H2O物質的量比為2∶2時的樣品S-1,可逆性優于其他樣品。
組裝的模擬電池在不同電流下的恒流放電曲線見圖4。
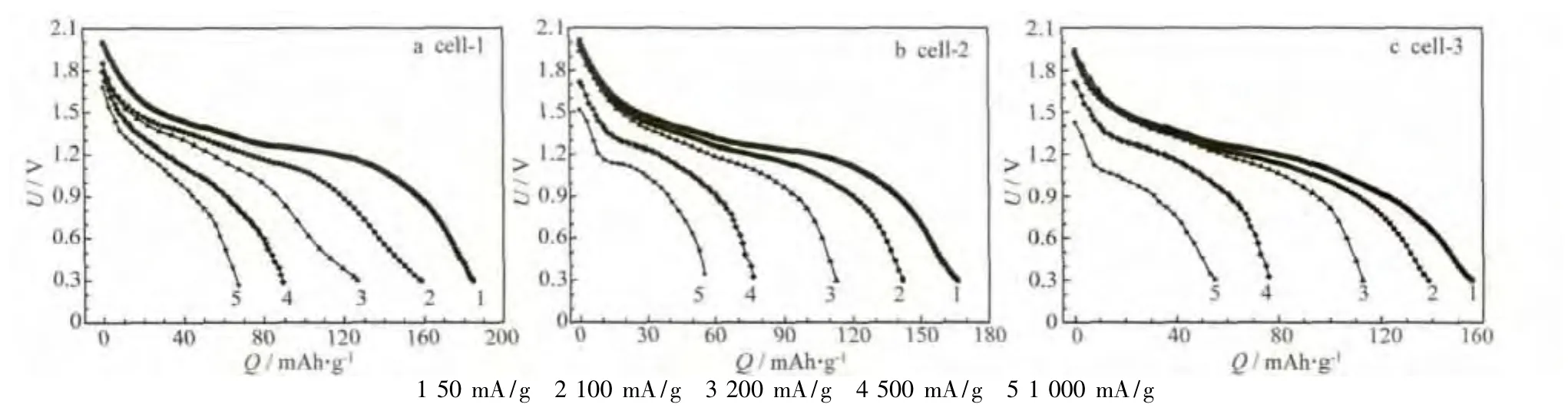
圖4 組裝的模擬電池的恒流放電曲線Fig.4 Galvanostatic discharge curves of assembled simulating cells
從圖4可知,當放電電流為50 mA/g時,cell-1、cell-2和cell-3的首次放電比容量分別為184 mAh/g、166 mAh/g和156 mAh/g。當放電電流為 1 000 mA/g時,cell-1、cell-2和cell-3的放電比容量分別為67 mAh/g、58 mAh/g和55 mAh/g,分別是50 mA/g時的36.35%、35.0%和35.38%,即放電電流增大20倍,放電比容量衰減了近2/3,說明各材料在高倍率放電下的容量下降程度差別不明顯。樣品S-1具有更好的電化學性能,是因為MnCl2·4H2O的增加會提高制備的納米棒狀α-MnO2的結晶度,棒狀結構的長徑比增大,不利于Zn2+在隧道中的傳輸,影響鋅離子電池的基礎反應。
組裝的模擬電池在首次放電后,在電流為100 mA/g時的恒流充放電循環曲線見圖5,循環性能見圖6。

圖6 組裝的模擬電池在電流為100 mA/g時的循環性能Fig.6 Cycle performance of assembled simulating cells at the current of 100 mA/g
從圖5、圖6可知,模擬電池在初始的幾次循環過程中,庫侖效率有波動,放電容量呈先升、后降的趨勢。前幾次循環為電極的活化過程,電池的比容量有一定的增加,cell-1、cell-2和cell-3的最高比容量分別為143 mAh/g、131 mAh/g和124 mAh/g。第50次循環時,cell-1、cell-2和cell-3的放電比容量分別衰減到110 mAh/g、104 mAh/g和92 mAh/g,容量保持率分別為76.8%、79.3%和74.0%。
3 結論
以KMnO4和MnCl2·4H2O為原料,采用低溫固相法制備直徑為7~10 nm、長度為50~120 nm的納米棒狀α-MnO2,樣品的晶型結構較純。隨著KMnO4與MnCl2·4H2O物質的量比的提高,粉體的結晶性提高、納米棒的直徑和長徑比減小。制備的鋅離子電池在電流為100 mA/g時的比容量最高為143 mAh/g,第50次循環的容量保持率為76.8%。隨著KMnO4與MnCl2·4H2O物質的量比的減小,組裝電池的比容量有所下降,但對循環性能的影響不大。
[1]XIA Xi(夏熙).二氧化錳及相關錳氧化合物的晶體結構制備及放電性能[J].Battery Bimonthly(電池),2004,34(6):412 -414.
[2]ZHANG Wei(張偉),LIU Kai-yu(劉開宇),ZHANG Ying(張瑩).α-MnO2的機械化學法合成及其電容性質[J].China’s Manganese Industry(中國錳業),2008,26(2):40 -52.
[3]XUE Zhao-hui(薛兆輝),LIU Zhao-lin(劉兆臨),MA Fang-wei(馬方偉),et al.水熱法合成α-MnO2納米棒及其電化學性能[J].Chinese Journal of Inorganic Chemistry(無機化學學報),2012,28(4):692 -696.
[4]WANG Xing-yan(汪形艷),WANG Xian-you(王先友),YANG Hong-ping(楊紅平),et al.超級電容器電極材料納米α-MnO2的制備及性能研究[J].Natural Science Journal of Xiangtan University(湘潭大學自然科學學報),2004,26(3):88-90.
