Castleman病繼發副腫瘤性天皰瘡和系統性紅斑狼瘡一例
2014-12-04 01:44:21吳瑋范瑞強羅家勝秦曉民
中華皮膚科雜志 2014年10期
吳瑋 范瑞強 羅家勝 秦曉民
Castleman病繼發副腫瘤性天皰瘡和系統性紅斑狼瘡一例
吳瑋 范瑞強 羅家勝 秦曉民
患者男,47歲。反復口腔潰瘍1年余,全身水皰9個月余。體檢:口腔內見多處潰瘍面,唇黏膜見片狀糜爛面、黃色或黑褐色痂皮和嘴角浸漬發白,張口困難;鼻孔黏膜交界處、眼瞼緣和陰莖冠狀溝見黃褐色痂和脫痂后的淡紅斑;軀干、四肢見散在紅斑和水皰,大皰皰壁緊貼皮膚、部分破裂,基底潮紅,類似多形紅斑樣改變,部分皰壁渾濁,見白色藥痂黏著或黑色痂皮,尼氏征陰性;10指(趾)甲周見皺縮的水腫性暗紅斑。水皰組織病理和皮損直接免疫熒光檢查符合天皰瘡診斷。縱隔腫瘤組織病理和免疫組化符合Castleman病(透明血管型)伴濾泡樹突細胞增生。實驗室檢查:血清補體C3下降以及抗核抗體、抗核小體抗體、dsDNA抗體均陽性。診斷: Castleman病、副腫瘤性天皰瘡、系統性紅斑狼瘡。用小劑量潑尼松和硫唑嘌吟治療2個月后皮疹完全消退,免疫抑制藥物緩慢減量,治療6個月后,患者自行停用免疫抑制藥物改為中藥治療,皮疹發展為中毒性表皮壞死松解癥,最后死亡。該病在腫瘤切除后免疫抑制藥物需維持一段較長時間,否則可導致皮疹反復或加重。
巨淋巴結增生;副腫瘤性天皰瘡;紅斑狼瘡,系統性
Castleman病是一種良性的淋巴細胞增生性腫瘤,又稱巨大淋巴濾泡增生,可引起特征性黏膜及皮膚損害,表現與天皰瘡相似的病理和免疫熒光,也可出現結締組織病的一系列表現。現報告 1例 Castleman病繼發副腫瘤性天皰瘡(paraneoplastic pemphigus)和系統性紅斑狼瘡(SLE)。
一、病歷資料
患者男,47歲,反復口腔潰瘍1年余,全身反復水皰9個月余。患者于1年前發熱后口腔出現多處疼痛性潰瘍,診治不詳,無好轉,2個月后龜頭上也出現糜爛和疼痛,曾到某口腔醫院診治,診斷為白塞病,治療后(具體不詳)龜頭糜爛好轉,但口腔潰瘍仍反復。就診前9個月,雙手掌出現散在的紅斑和水皰,伴瘙癢,之后四肢和軀干多處出現紅斑和水皰,瘙癢劇烈。患者一直用中藥煎劑治療,病情反復。就診前20天皮疹增多,全身出現水皰、糜爛和結痂,口腔潰瘍加重,逐漸波及到雙唇、鼻黏膜和眼瞼緣,潰瘍表面結痂,張口受限,遂到我院,查血常規、Trust試驗、HIV抗體均正常,予抗過敏藥物效果不佳。擬診天皰瘡于2012年8月18日收入我科。發病以來患者體重減輕10 kg,無關節疼痛和發熱,家族中無類似病史。既往史:否認高血壓、糖尿病、腎病、肝炎、肺結核等病史,否認藥物過敏史。5年前曾行胸部CT檢查提示右上縱隔一直徑約2 cm的腫物,考慮為腫大淋巴結,未作處理。
體檢:體溫37℃,脈搏80次,呼吸24次,血壓120/80 mmHg (1 mmHg=0.133 kPa)。精神疲憊,消瘦。全身淺表淋巴結未及腫大,心肺腹無明顯異常。皮膚科情況:雙側頰黏膜、上顎黏膜、舌邊緣見多處潰瘍面;唇黏膜見片狀糜爛面、滲出、黃色或黑褐色痂皮和嘴角浸漬發白,張口困難;鼻孔黏膜交界處、眼瞼緣和陰莖冠狀溝見漿痂和脫痂后的淡紅斑;軀干、四肢見散在紅斑和水皰,大皰皰壁緊貼皮膚、部分破裂,基底潮紅,類似多形紅斑樣改變,部分皰液渾濁,見白色藥痂黏著或黑色痂皮,尼氏征陰性;10指(趾)甲周見皺縮的水腫性暗紅斑。見圖1~3。
實驗室檢查:白細胞5.5×109/L,嗜酸性粒細胞0.12、計數1×109/L,血紅蛋白119 g/L(參考值135~175 g/L)。免疫功能6項(2次結果相似):IgM 2.48 g/L(0.4~2.3 g/L),C3 0.43 g/L(0.9~1.8 g/L)、CH50 20 U/m(l23~46 U/ml),C4正常。自身免疫抗體(2次結果相似):抗ANA抗體1∶1 000、均質型,抗組蛋白抗體弱陽性,抗核小體抗體陽性,抗dsDNA抗體(++),重組Ro-52弱陽性。總IgE 632.4 IU/ml (0~200 IU/ml);EB病毒抗體VCA-IgA陽性,EB病毒早期抗體EA-IgA陰性。血細胞沉降率19 mm/1 h;白蛋白31.4 g/L (40~55 g/L)。尿糞常規、肝功能、凝血4項、肺癌3項、前列腺癌標抗體、血管炎3項(抗PR3、MPO、GBM抗體)、CA242、CA199、癌胚抗原、甲胎蛋白、類風濕因子、C反應蛋白均正常。結核抗體、天皰瘡抗體和類天皰瘡抗體均陰性。X線胸片:右上縱隔腫塊5 cm×6 cm×7 cm。胸部CT(圖4):右前上縱隔內見一團狀軟組織腫塊影,邊界清楚,大小7.2 cm× 5.9 cm×9.6 cm,考慮:右前縱隔內腫塊,巨淋巴結增生可能,心包少許積液;雙肺散在少許炎癥,雙肺上葉多發性肺大泡。背部水皰組織病理檢查(圖5):輕度角化過度,部分基底細胞層上水皰,部分為棘層內水皰,水皰內見棘刺松解細胞,真皮淺層及血管周圍散在淋巴細胞和嗜酸性粒細胞浸潤;直接免疫熒光:棘細胞間IgG(±)、C3(++)(圖6),基底膜帶IgG(+), IgA和IgM均為陰性。符合天皰瘡組織病理改變。
二、治療經過
根據皮損組織病理和免疫熒光,結合臨床和實驗室檢查,擬診副腫瘤性天皰瘡、Castleman病?SLE。給予丙種球蛋白5 g靜脈注射每日1次,用5 d,潑尼松25 mg/d,于同年9月12日在胸外科行右上前縱隔腫物切除術,并清除頸部淋巴結20個。縱隔腫物病理檢查:包膜完整,表面灰紅,質地細膩。腫瘤組織中可見結節狀成分和彌漫生長成分:結節狀成分為生發中心萎縮伴透明變的淋巴濾泡,周邊套區小淋巴細胞增生,圍繞生發中心呈同心圓樣排列,可見毛細血管穿透套區并長入生發中心(圖7);彌漫生長區由梭形至卵圓形細胞構成,排列呈束狀、席紋狀,瘤細胞核卵圓形或長梭形,核仁小,核膜薄,瘤細胞異形性不明顯,梭形細胞間見均勻粉染沉積,間質淋巴細胞、漿細胞及組織細胞反應明顯,局部見多核巨細胞。免疫組化結果:CD3、CD5、CD7、CD20、PaX-5、CD21、CD23均陽性,CD15、CD30、TdT、SMA、CD35均陰性;散在漿細胞CD38、CD138陽性,血管CD34陽性;符合Castleman病(透明血管型)伴濾泡樹突細胞增生及間質硬化。另見淋巴結20個,呈反應性增生改變。最后確診為Castleman病(透明血管型)、副腫瘤性天皰瘡、SLE。術后繼續予潑尼松25 mg每日1次,4周后皮膚糜爛面大部分消退,腋下和胸壁仍有水皰和糜爛面,唇黏膜和口腔內仍有糜爛和疼痛,影響進食,加服硫唑嘌吟50 mg每日2次,治療4周后皮疹和黏膜糜爛基本消失。12月6日自身抗體結果:ANA 1∶320均質型,抗組蛋白抗體、抗核小體抗體、抗dsDNA、重組Ro-52均陰性;IgM 3.51 g/L,C3 0.61 g/L、CH50、C4正常。2013年1月,復查胸部CT未見異常,黏膜潰瘍和皮疹無復發,糖皮質激素和免疫抑制劑逐漸減量。同年3月,患者突然自行停用潑尼松和硫唑嘌吟,改為純中藥治療,1周后口腔潰瘍復發,全身出現散在水皰,堅持僅服中藥治療,1個月后出現中毒性表皮壞死松解癥樣皮疹。胸片:右上肺前段、右中肺纖維灶,未除外左肺上葉支氣管擴張并感染,未見縱隔腫物。自身抗體: ANA 1∶3 200,dsDNA(-),補體C3 0.51。最后死于感染性多器官功能衰竭。

圖1 患者唇黏膜見糜爛面、滲出、黃色或黑褐色痂皮,口角浸漬發白
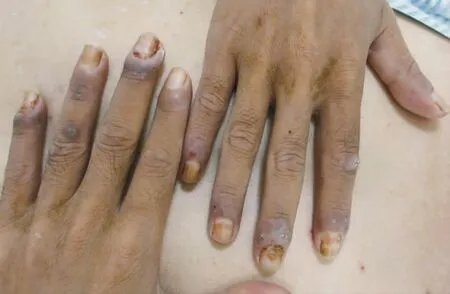
圖2 10指甲周見皺縮的水腫性暗紅斑
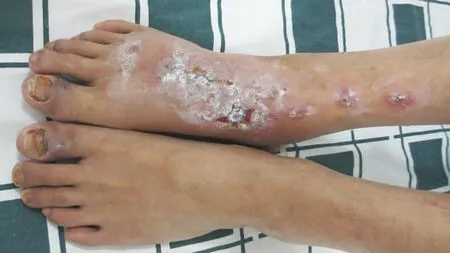
圖3 趾甲周見水腫性暗紅斑,右足背和小腿暗紅斑,見糜爛面和水皰

圖4 胸部CT:右前上縱隔內見團狀軟組織腫塊影,邊界清楚
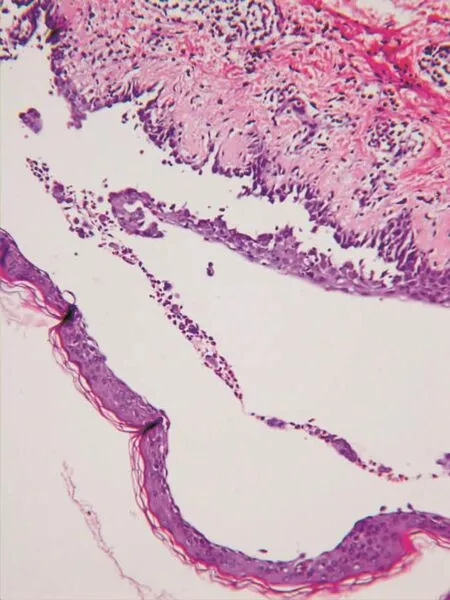
圖5 皮損組織病理:表皮內水皰,棘層松解細胞,真皮淺層及血管周圍散在淋巴細胞和嗜酸性粒細胞浸潤(HE×200)

圖6 直接免疫熒光:C3棘細胞間沉積(HE×200)

圖7 小血管長入生發中心,內皮細胞增生,生發中心透明變,周圍小淋巴細胞呈洋蔥皮樣排列(HE×200)
三、討論
Castleman病是一種病因不明的良性淋巴結增生性疾病,可累及淋巴結區域的任何部位,以胸部縱隔淋巴結最常見,其次是頸部、腋下、腹部和腹股溝等部位。該患者5年前行胸部CT曾發現右上縱隔處有1處直徑約2 cm的腫大淋巴結,逐漸增大到直徑6 cm,說明Castleman病是一個良性增生過程,對于無癥狀的深部腫大淋巴結是否需早期切除是個值得探討的問題。根據組織病理學Castleman病分為3種類型:透明血管型、漿細胞型、混合型。本例患者根據臨床表現、胸部CT和病理結果提示為單中心透明血管型Castleman病[1]。Castleman病的治療方法和療效與其分型和繼發的疾病有關,對于單中心型首選手術切除腫瘤。2005年朱學駿等[2]報告對于伴發副腫瘤性天皰瘡的單中心型Castleman病患者早診斷、早切除腫瘤是成功治療副腫瘤性天皰瘡的關鍵。本例患者也采取了相同的治療方法,并在縱隔腫瘤切除后取得較好的短期療效,但用小劑量糖皮質激素和硫唑嘌吟治療6個月后患者突然停用轉而改服中藥煎劑,出現中毒性表皮松解癥樣皮疹,最后死亡。
副腫瘤性天皰瘡并發的腫瘤很多,在中國以Castleman病為最常見[3]。王亮春等[4]對Castleman腫瘤組織細胞進行培養及鑒定培養細胞上清液中抗體成分,發現Castleman腫瘤組織能夠分泌抗體,且抗體性質與血液中的抗體性質基本相同。Tey等[5]曾報道Castleman病可能始發于暴露于表皮表位的界面皮炎,從淋巴腫塊釋放出來的IL-6激活了分子模擬和表位擴展,導致自身抗體的產生。皮損形態的多樣性可能與疾病發展過程中以細胞免疫還是體液免疫介導為主有關,各種皮疹可同時共存,也可由一種皮疹發展成另一種皮疹,當表現為松弛性大皰時類似尋常型天皰瘡和落葉型天皰瘡,這些大皰可能發展為大面積表皮剝脫,類似于中毒性表皮壞死松解癥[6]。Kim等[7]報道1例患SLE1年的41歲患者,出現難治性口腔潰瘍和軀干部暗紅斑,皮膚病理活檢提示天皰瘡樣改變,影像學檢查發現腹膜后巨大腫塊,病理活檢提示為透明血管型Castleman病,切除腫瘤后SLE的自身抗體組蛋白抗體轉陰,ds-DNA抗體明顯下降,與本例非常相似。
總之,需要強調的是,Castleman病導致副腫瘤性天皰瘡和SLE,與結締組織病之間的因果關系還需進一步研究,同時強調與Castleman病相關的副腫瘤性天皰瘡在腫瘤切除后,免疫抑制藥物應用需維持一段很長的時間,否則容易導致皮疹的反復和加重。
志謝廣州醫學院附屬第一醫院病理科付欣鴿教授
[1]Wen X,Jiang X.Paraneoplastic pemphigus in association with Castleman disease of the pararenal retroperitoneum[J].J Dermatol,2012,39(7):662-664.
[2]朱學駿,王京,陳喜雪,等.伴發副腫瘤性天皰瘡的Castleman瘤:附10例報告[J].中華皮膚科雜志,2005,38(12):745-747.
[3]Zhu X,Zhang B.Paraneoplastic pemphigus[J].J Dermatol,2007,34(8):503-511.
[4]王亮春,陳喜雪,趙俊郁,等.副腫瘤性天皰瘡伴發Castleman腫瘤分泌致病相關抗體的初步研究[J].中華皮膚科雜志,2004,37(2):74-76.
[5]Tey HL,Tang MB.A case of paraneoplastic pemphigus associated with Castleman′s disease presenting as erosive lichen planus[J]. Clin Exp Dermatol,2009,34(8):e754-e756.
[6]Yong AA,Tey HL.Paraneoplastic pemphigus[J].Australas J Dermatol,2013,54(4):241-250.
[7]Kim KJ,Cho CS,Choi JJ.Pararenal retroperitoneal Castleman′s disease mimicking systemic lupus erythematosus[J].Int J Rheum Dis,2010,13(3):e20-e25.
2014-03-10)
(本文編輯:吳曉初)
Castleman′s disease with secondary paraneoplastic pemphigus and systemic lupus erythematosus:a case report
Wu Wei,Fan Ruiqiang,Luo Jiasheng,Qin Xiaomin.Department of Dermatology,Guangdong Provincial Hospital of Traditional Chinese Medicine,Guangzhou 510120,China
Wu Wei,Email:wuwei1350187@126.com
A 47-year-old man presented with recurrent oral ulcerations for more than 1 year and generalized vesicles for 9 months.Physical examination revealed multiple oral ulcers,patchy labial erosions with yellow or dark brown crusts,maceration and blanching of the angle of mouth resulting in difficulty in mouth opening. Cinnamomeous crusts were seen on the nasal limen,palpebral margin and coronary sulcus of penis,with pale red macules after decrustation.Diffuse erythematous patches and bullae were present on the trunk and extremities.The walls of bullae tightly clung to the skin,and some bullae were ruptured,leaving an erythematous and moist surface and giving an erythema multiforme-like appearance;some bullae were turbid and covered with adherent drugcontaining white crusts or black crusts.Nikolsky′s sign was negative.Shrinking,edematous dark-red patches were seen in periungual regions of all the fingers and toes.Histologic biopsy and direct immunofluorescence examination ofbullous lesions confirmed a diagnosis ofparaneoplastic pemphigus (PNP).Histopathologicaland immunohistochemical findings from the mediastinal tumor were consistent with Castleman′s disease(hyalinevascular type)with proliferation of follicular dendritic cells.Laboratory examination revealed a decrease in serum complement C3 and the presence of antinuclear,anti-nucleosome and anti-dsDNA antibodies.The final diagnosis included Castleman′s disease,PNP and systemic lupus erythematosus.After 2 months of treatment with low-dose prednisone and azathioprine,skin lesions completely regressed.Then,the dose of prednisone and azathioprine was tapered.Six months later,the patient himself suddenly withdrew prednisone and azathioprine,and began to take traditional Chinese medicine;thereafter,the lesions developed into toxic epidermal necrolysis(TEN),and the patient died finally.This case demonstrates that immunosuppressive therapy should be maintained for a long period of time in patients with Castleman′s disease after tumor removal,otherwise,skin lesions may recur or get worse.
Giant lymph node hyperplasia;Paraneoplastic pemphigus;Lupus erythematosus,systemic
10.3760/cma.j.issn.0412-4030.2014.10.020
510120廣州,廣東省中醫院皮膚科
吳瑋,Email:wuwei1350187@126.com
