紅花藥材中非法染有酸性紅73的TLC鑒別和HPLC確證
2014-09-26 07:45:07楊榮艷宋瑩李本淳
中國現代中藥 2014年8期
關鍵詞:方法
楊榮艷,宋瑩,李本淳
(吉林省四平市食品藥品檢驗所,吉林 四平 136000)
紅花藥材中非法染有酸性紅73的TLC鑒別和HPLC確證
楊榮艷*,宋瑩,李本淳
(吉林省四平市食品藥品檢驗所,吉林 四平 136000)
目的:建立紅花藥材中非法染有酸性紅73的檢測方法。方法:采用TLC對紅花中非法染有酸性紅73進行定性鑒別;采用HPLC確證建立的TLC可行。結果:在TLC中,陽性樣品可檢出與酸性紅73對照試劑位置和顏色一致的斑點;HPLC中,也出現了與對照試劑保留時間一致的色譜峰,并且DAD檢測器檢驗,在420~580 nm波長范圍的紫外-可見吸收光譜相同。結論:該方法前處理簡便、快速、準確,可用于紅花藥材中非法染有酸性紅73的檢查。
紅花;酸性紅73;薄層色譜法;高效液相色譜法
紅花收載于《中國藥典》2010年版一部,檢驗項目有性狀、顯微鑒別、薄層鑒別、水分檢查、雜質檢查、總灰分檢查、酸不溶性灰分檢查、吸光度檢查、浸出物和含量測定[1]。《藥品檢驗補充檢驗方法和檢驗項目批準件(編號2007009)》[2]中補充了紅花金橙Ⅱ檢查項。在2012年度藥品抽驗中,筆者按照《中國藥典》2010年版一部對20批紅花進行薄層鑒別檢驗時,發現有3批紅花供試品色譜比對照藥材色譜相應的位置上多一個紅色斑點,并對此斑點的成分進行了研究,初步確認為酸性紅73。酸性紅73是一種強烈的致癌物質,被國家禁止用于食品中的工業染料,威脅患者的健康[3]。筆者通過實驗建立薄層色譜定性鑒別和高效液相色譜確證紅花中非法染有酸性紅73的方法,為藥品監管提供技術支持。
1 儀器與試藥
1.1 儀器
LC-2010CHT型高效液相色譜儀(島津),紫外檢測器和DAD檢測器,CLASS-VP色譜工作站,UV-2450可見-紫外分光光度計,BP211D型電子分析天平,LC-250超聲清洗器(功率250 W,頻率50 kHZ);純水器(德國)。
1.2 試藥
酸性紅73對照試劑(美國AccuStandard公司,供含量測定用,批號:16675);色譜甲醇,自制超純水,醋酸銨、乙醇均為分析純,市售硅膠G板(上海盛亞化工有限公司,批號:20090306)。
紅花(市售),紅花對照藥材(中國食品藥品檢定研究院,批號:120907-201111)。
2 方法與結果
2.1 薄層色譜鑒別[4]
取本品1 g,加70%乙醇20 mL,超聲提取20 min,取上清液作為供試品溶液。另取酸性紅73對照試劑適量,分別加70%乙醇溶解并制成每1 mL含60 μg的溶液,作為對照試劑溶液。照《中國藥典》2010年版一部薄層色譜法(附錄Ⅵ B)試驗,吸取供試品溶液和對照試劑溶液各5 μL,分別點于同一硅膠G薄層板上,以乙酸乙酯-正丁醇-乙醇-氨水-水(1∶3∶3∶1∶1)為展開劑,展開,取出,晾干,在可見光下檢視。供試品色譜中,在與對照試劑色譜相應的位置上,不顯相同顏色的斑點;陰性對照不干擾測定。見圖1。
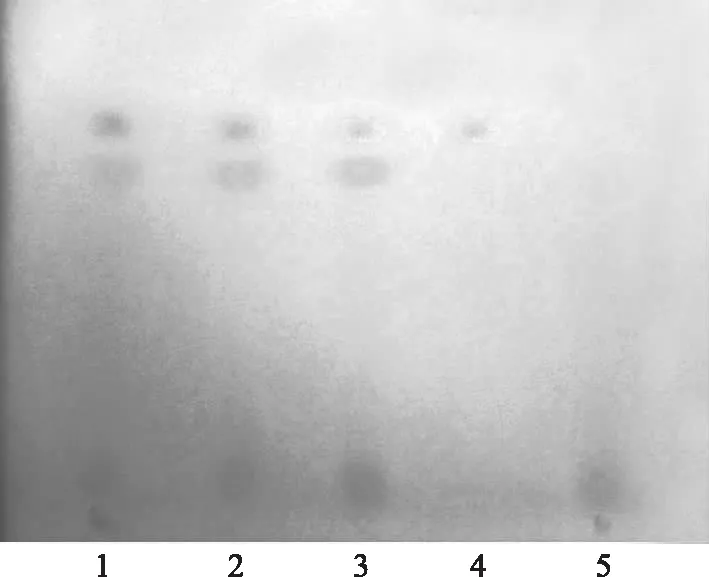
1~3.供試品 4.酸性紅73對照試劑 5.紅花對照藥材圖1 紅花及對照品TLC圖
2.2 HPLC測定酸性紅73(紫外檢測器)
2.2.1 色譜條件[5-6]色譜柱:Agilent TC-C18(250 mm×4.6 mm,5 μm),流動相:甲醇-0.025 mol·L-1醋酸銨溶液,甲醇梯度洗脫條件:20%~45%(0~8 min),45%~80%(8~20 min);流速:1.0 mL·min-1,檢測波長:508 nm,進樣量:10 μL,柱溫:35 ℃。2.2.2 對照品溶液的制備 酸性紅73對照試劑12 mg,精密稱定,置200 mL量瓶中,加70%乙醇稀釋至刻度,即得(每1 mL含酸性紅73對照試劑60 μg)。
2.2.3 供試品溶液的制備[5,7]取本品約1 g,加70%乙醇20 mL,超聲處理20 min,放冷,取續濾液,即得。
2.2.4 陰性樣品溶液的制備 取不含有酸性紅73的紅花對照藥材作為陰性樣品,按照2.2.3供試品溶液的制備方法制備陰性樣品溶液。
2.2.5 專屬性試驗 把供試品溶液、陰性樣品溶液和酸性紅73對照試劑分別注入高效液相色譜儀,依法測定,結果供試品色譜圖中,供試品主峰保留時間與酸性紅73對照試劑主峰保留時間一致;陰性樣品色譜圖中,在與酸性紅73對照試劑主峰保留時間處無峰出現。見圖2。
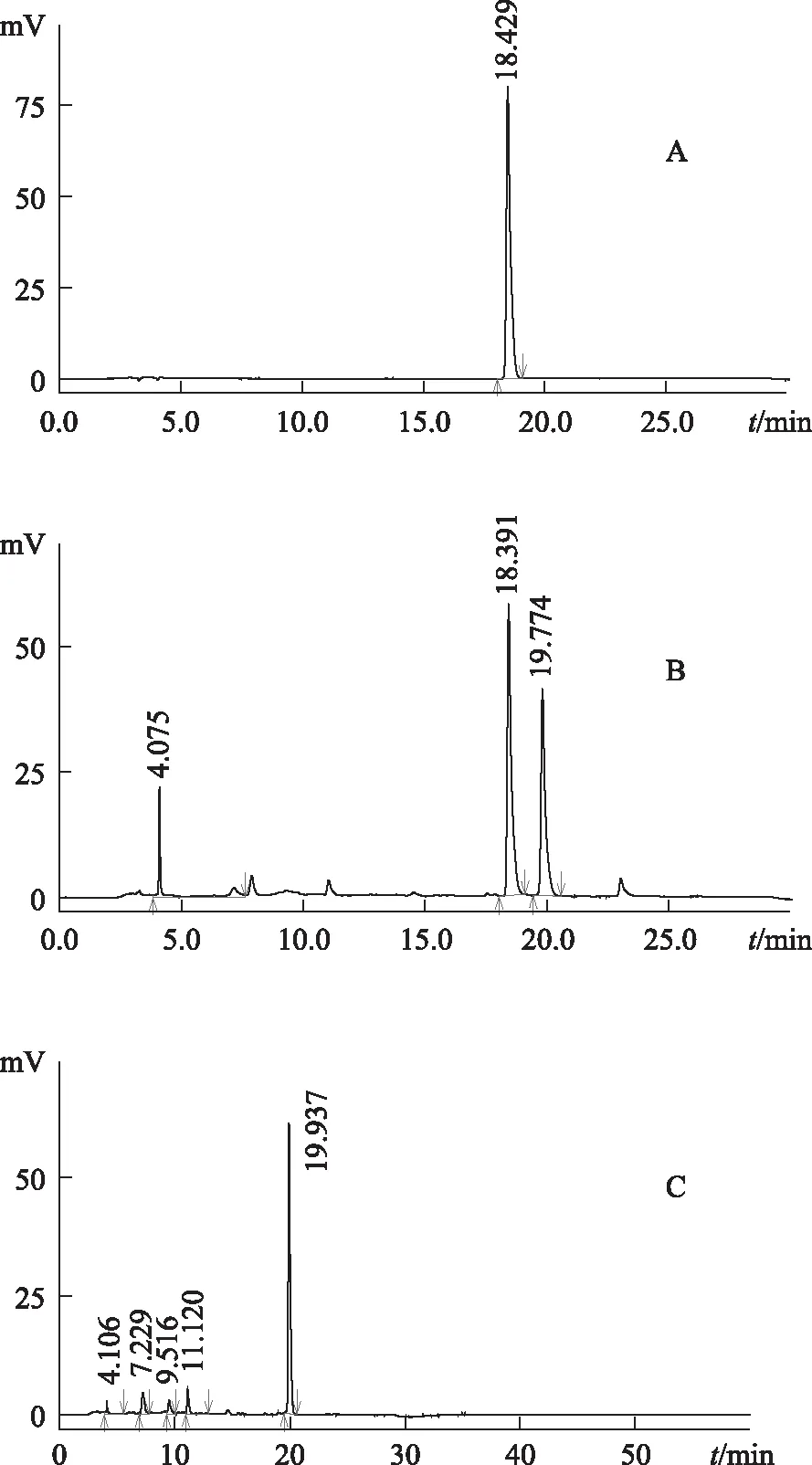
A.酸性紅73對照試劑 B.供試品 C.紅花對照藥材圖2 紅花及對照品HPLC圖
2.2.6 線性關系的考察 精密稱取酸性紅73對照試劑(含量測定用)10.10 mg,置100 mL容量瓶中,加用0.45 μm的微孔濾膜過濾后的70%乙醇適量,超聲溶解并定容至刻度,搖勻,作為對照試劑溶液儲備液。
精密量取對照試劑溶液儲備液0.1,0.5,1,2,3,6,8,10 mL分別置10 mL量瓶中,用上述70%乙醇定容至刻度,搖勻,即得質量濃度分別為1.01,5.05,10.10,20.20,30.30,60.60,80.80,100.10 μg·mL-1系列標準溶液。不經微孔濾膜過濾,分別將上述系列標準溶液各10 μL注入高效液相色譜儀分析測定,每個濃度連續進樣3次,取3次平均值,以質量濃度為橫坐標(X),峰面積為縱坐標(Y),經線性回歸,得回歸方程Y=30 393X-2 583.8,r=1.000。結果表明,酸性紅73在1.01~100.10 μg·mL-1具有良好的線性關系。
2.2.7 精密度試驗 取同一質量濃度(60 μg·mL-1)對照品溶液按上述色譜條件測定,連續進樣6次,結果峰面積RSD=0.34%(n=6),表明儀器精密度良好。
2.2.8 穩定性試驗 取同一供試品溶液,照上述色譜條件測定,考察了0,2,4,8,12,16,24 h峰面積值的變化,結果峰面積RSD=1.18%(n=6),表明樣品溶液在24 h內穩定。
2.2.9 重復性試驗 取同一供試品12份,共分為2組,每份1.0 g,精密稱定,精密加入70%乙醇20 mL,照2.2.3方法制備,獨立試驗,結果第一組中酸性紅73的平均質量分數為0.40 mg·g-1,RSD=5.12% (n=6);第二組中酸性紅73的平均質量分數為0.48 mg·g-1,RSD=8.03%(n=6)。
2.2.10 陰性樣品回收率試驗 稱取紅花對照藥材樣品6份,每份約0.5 g,精密稱定,分別置具塞錐形瓶中,分別精密加入對照試劑溶液10 mL(60 μg·mL-1),再分別精密加入70%乙醇10 mL,稱定重量,超聲處理20 min,放冷,補足減失的重量,搖勻,用0.45 μm的微孔濾頭(用待過濾液4 mL使濾頭充分飽和)過濾,進樣量10 μL,注入高效液相色譜儀測定,即得。測定結果見表1。
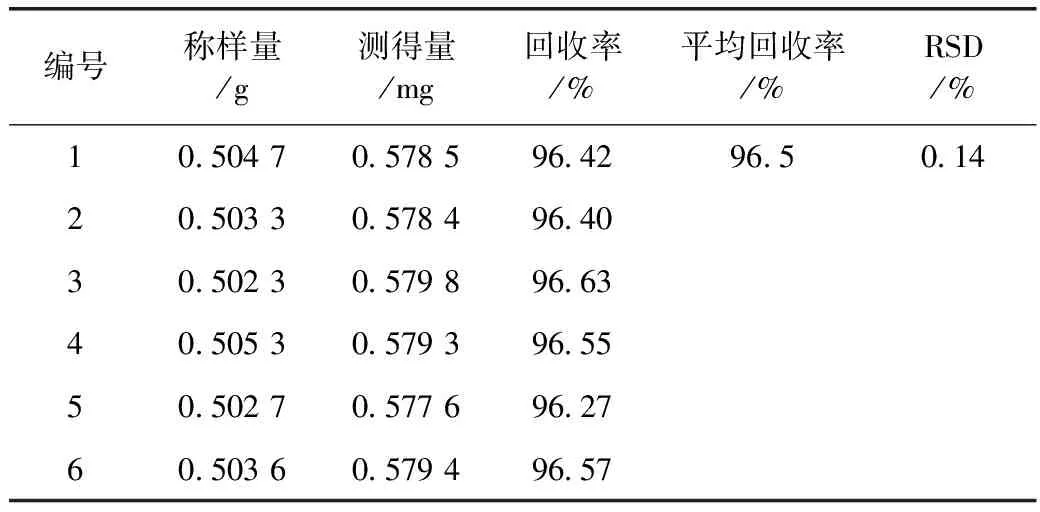
表3 紅花陰性樣品回收率試驗
注:樣品中含酸性紅73的量均為0 mg,酸性紅73對照試劑加入量均為0.600 0 mg
2.2.11 方法的檢測限 按照正文色譜條件,基線走穩后,進一針10 μL溶劑,再分別將質量濃度為0.1,0.2,0.3 μg·mL-1的對照試劑各10 μL注入高效液相色譜儀,結果0.3 μg·mL的對照試劑信噪比S/N=3,所以將方法的檢測限定為0.3 μg·mL-1。
2.2.12 樣品測定 照上述色譜條件和制備方法,依法測定3批市售紅花樣品,分別測定3次,3批樣品中酸性紅73的質量分數分別為0.45,0.71,0.86 mg·g-1。
2.3 高效液相色譜法DAD檢測器確證
2.3.1 色譜條件、供試品和對照品試劑的制備 DAD檢測器,其他條件同2.2.1;供試品和對照品試劑的制備分別同2.2.2和2.2.3。
2.3.2 高效液相色譜法DAD檢測器檢驗 供試品色譜中,出現與對照試劑保留時間一致的色譜峰。采用二極管陣列檢測器比較相應色譜峰在420~580 nm波長范圍的紫外-可見吸收光譜,發現吸收光譜相同。見圖3~6。
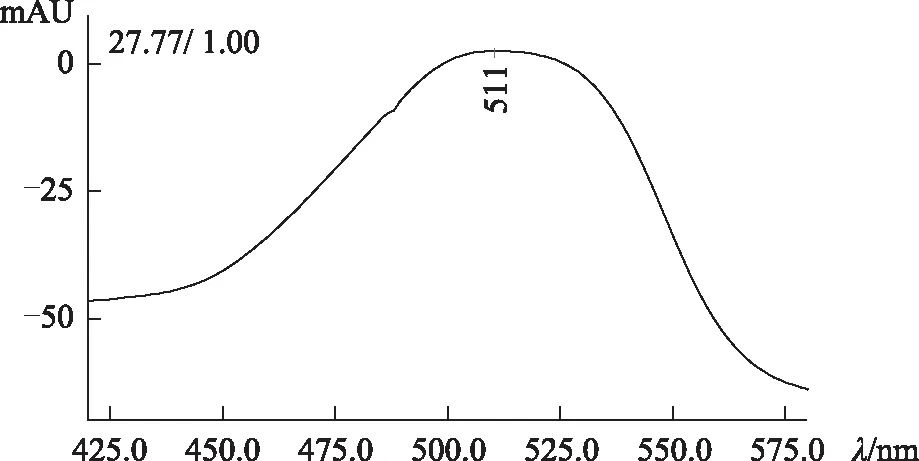
圖3 酸性紅73對照試劑光譜
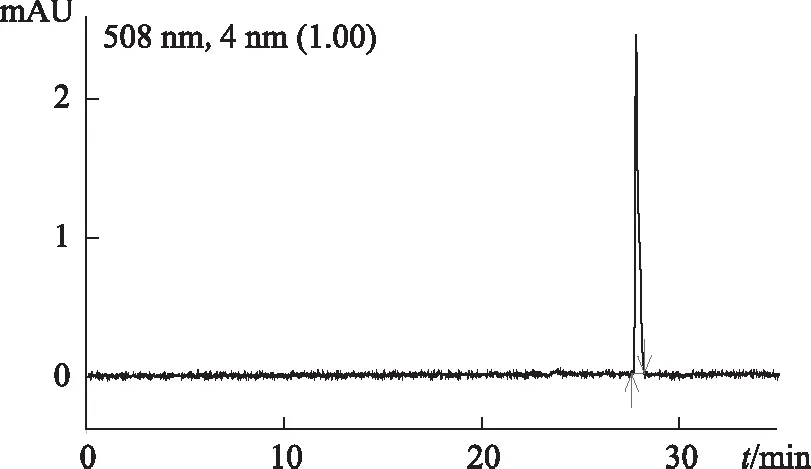
圖4 酸性紅73對照試劑色譜
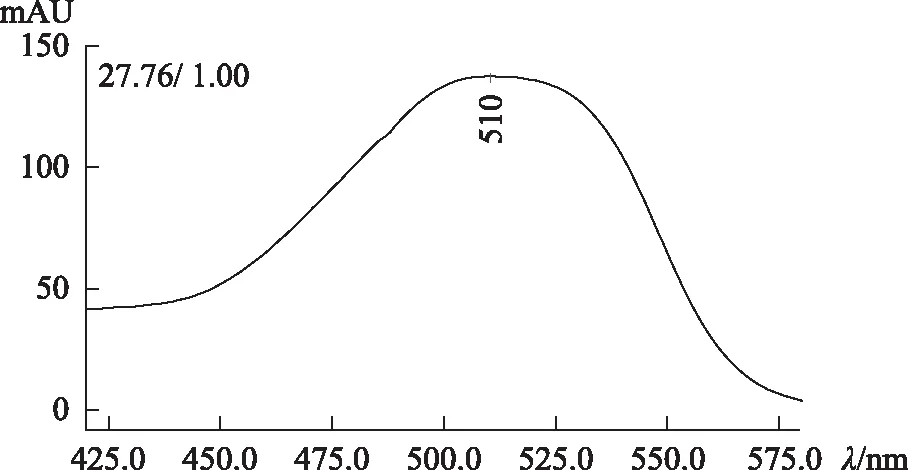
圖5 紅花供試品光譜
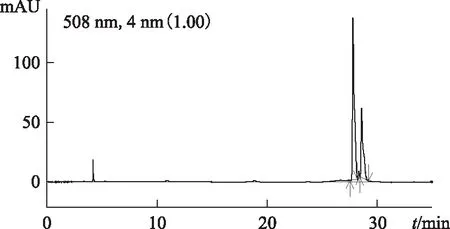
圖6 紅花供試品色譜
3 討論
實驗中發現兩組重復性試驗結果RSD值均大于3.0%,為此考察了樣品粉碎與否對結果的影響,結果發現不粉碎的樣品含量高于粉碎樣品的含量,樣品處理得越細,含量越低,而且含量也不均勻。考慮到酸性紅73是一種著色劑,一般情況下都會附著在樣品的表面,所以采取不粉碎處理樣品。
另外,實驗中發現不同濃度的對照試劑溶液經0.45 μm微孔濾頭過濾后,峰面積與濃度比不成正比,考慮到可能是微孔濾頭對酸性紅73具有吸附作用。我們采用同濃度對照試劑溶液,通過過濾和未過濾來驗證是否具有吸附作用,考察結果發現,未經過濾的對照試劑的峰面積是經過濾的對照試劑的峰面積的1.2倍,損失率約占16%。考慮到每次用濾頭過濾供試品溶液時,其對供試品溶液中酸性紅73吸附程度不同,從而導致供試品含量不確定。上述結果說明樣品本身的均勻性差和酸性紅73的吸附性是影響重復性試驗結果的原因。實驗方法只是針對非法染有色素酸性紅73的紅花樣品檢查,并非含量測定項,雖然RSD超出3.0%,但并不影響方法的應用。
通過12份樣品的重復性試驗發現,紅花樣品中酸性紅73的質量分數在0.38~0.52 mg·g-1,表明樣品本身的均勻性很差,如采用樣品加標,無法得到準確的回收率結果,所以我們采取陰性樣品加標進行回收試驗,結果RSD=0.14%,符合《中國藥典》規定的要求,說明此檢驗方法可行。
考察了shimpack C18、zorbax SB C18、Agilent TC-C183種色譜柱,結果都能得到很好的色譜分離。另外,考察了流動相中醋酸銨溶液的兩種濃度即0.025,0.05 mol·L-1,結果都能達到系統適用性要求,但是考慮到含高濃度鹽的流動相對色譜柱傷害較大,所以采用0.025 mol·L-1醋酸銨溶液。
實驗建立的檢驗方法前處理簡便、快速、準確,可用于紅花藥材中非法染有酸性紅73的檢查,研究為紅花監管提供技術支持。
[1] 國家藥典委員會.中國藥典[S].一部.北京:中國醫藥科技出版社,2010:141-142.
[2] 國家食品藥品監督管理局.藥品檢驗補充檢驗方法和檢驗項目批準件匯編(2003~2008年)[S].2007:154.
[3] 國家食品藥品監督管理局稽查局.五味子補充檢驗方法和檢驗項目批準件[S].2007
[4] 閔春艷,付凌燕,汪祺,等.紅花藥材摻偽染色檢測方法的實驗研究[J].中國藥事,2011,(8):772-775.
[5] 肖海龍,屠海云,王紅青,等.反相高效液相色譜法快速測定食品中18種水溶性合成著色劑[J].中國衛生檢驗雜志,2011,21(2):264-266.
[6] 鄒耀華,殷紅妹,郭怡飚,等.HPLC-PDA法檢測西紅花和紅花中十一種非法添加色素[J].中國衛生檢驗雜志,2010,(11):2724-2725.
[7] 汪建君,陳惠玲.HPLC-PDA法檢測正天丸和正天膠囊中6種非法添加色素[J].藥物評價研究,2013,(1):51-53.
TLCHPLCIdentificationofillegallystainedAcidRed73inCarthamiFlos
YANG Rongyan*,SONG Ying,LI Benchun
(SipingInstituteforDrugandFoodControl,Siping136000,China)
Objective:To establish TLC identification and HPLC confirmatory method for illegally stained Acid Red 73 in Carthami Flos.Methods:TLC was employed to identify Acid Red 73,which was confirmed with HPLC.Results:In TLC,the emergence of Acid Red 73 with a consistent position and color spots.In HPLC,the emergence of the peak retention time,and comparing the wavelength range of 420-580 nm UV-visible absorption spectrum by a DAD detector,the same absorption spectrum was found.Conclusion:The method is easy,quick and accurate,can be used to identify Acid Red 73 in Carthami Flos.
Carthami Flos;Acid Red 73;TLC;HPLC
10.13313/j.issn.1673-4890.2014.08.005
2013-09-12)
*
楊榮艷,碩士,主管藥師,研究方向:中藥檢驗;E-mail:yry_lg@126.com
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
