阻斷p38絲裂原活化蛋白激酶在細胞空泡形成中的作用*
2014-09-22 07:48:42張春燕馮春紅敬健雄段春燕劉友平夏先明代榮陽陳紹坤
重慶醫學 2014年22期
張春燕,馮春紅,敬健雄,段春燕,劉友平,夏先明,李 洪,代榮陽,陳紹坤
(瀘州醫學院:1.生物化學教研室;2.肝膽外科;3.生物教研室,四川瀘州646000)
為了適應環境變化,細胞會發生一系列諸如皺縮、膨脹、分裂和空泡形成等形態學的改變。化學和生物活性物質誘導細胞形成的細胞質空泡是一種顯著且常見的現象[1-2],此外,細胞空泡也能自發形成[3]。細胞空泡化的程度主要取決于細胞類型[4-5],有些細胞極易發生空泡化,有些細胞則很難形成空泡。
有報道表明[6-7],細胞空泡就好比是細胞內幫助消化的酸性區域。這個酸性區域的組成、來源及形成機制除了取決于細胞類型,也取決于細胞空泡的誘導劑。有學者認為[8-9],細胞的自體吞噬介導了空泡形成,細胞空泡可能代表了某種典型的自體吞噬。但是,有的細胞空泡并非來自于自體吞噬,自體吞噬也并非總 是 介 導 空 泡 形 成[10-11]。 也 有 學 者 認 為[12-13],細 胞 空泡與細胞凋亡密切相關。有些凋亡誘變劑可誘導細胞空泡化,并通過空泡形成參與細胞凋亡的調控。值得注意的是[14],有的細胞空泡并不是因凋亡而產生,有些空泡誘導劑誘導了細胞空泡形成,但并不引發細胞凋亡。絲裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK),如p38MAPK是細胞內重要信號通路,MAPK通路在細胞炎癥反應、凋亡、生長和細胞應激等多種生理和病理過程中起著重要的調節作用。雖然目前已明確細胞空泡與細胞應激反應密切相關[15],但是,細胞空泡的形成是否受到細胞應激反應信號通路p38MAPK的調節尚不清楚。
本實驗旨在探討p38MAPK是否參與了細胞空泡形成。研究發現細胞空泡可在HepG2細胞中自發形成,這些空泡可被茴香霉素(Anisomycin)通過活化p38MAPK而消除。此外,阻斷p38MAPK可誘導多種腫瘤細胞空泡形成。本實驗結果表明p38MAPK在調節細胞空泡形成中發揮了重要作用。
1 材料與方法
1.1 材 料 Anisomycin、放 線 菌 酮 (Cycloheximide,CHX)、SB203580、SP600125購自Merck公司;細胞培養試劑購自Gibco公司;β-actin、p-MAPKAPK2、MAPKAPK2、p38MAPK、磷酸化c-Jun抗體購自CST公司;內質網紅色熒光探針及溶酶體紅色熒光探針購自Invitrogen公司;電泳試劑均購自Bio-Rad公司;其他為國產分析純。
1.2 實驗方法
1.2.1 細胞培養及處理 腫瘤細胞株 HepG2、LM3、QBC939、Hela和A549于37℃、5%CO2培養箱中常規培養,選取對數生長期細胞進行后續實驗。培養細胞分別用anisomycin(0.25、0.5、1.0μg/mL)、SB203580(0、2.5、5.0、10.0 μmol)、SP600125(20nmol)及CHX(5、10、15μmol)處理,并按在對應時間點觀察細胞空泡化情況。
1.2.2 Western blot法檢測p38MAPK等通路相關分子的表達水平 HepG2細胞常規培養,各實驗組給予不同的處理因素后,收集細胞,提取各組細胞蛋白,進行 Western blot檢測。總蛋白經十二烷基磺酸鈉-聚丙烯酰氨凝膠電泳(SDS-PAGE)分離后,轉移到PVDF膜上,脫脂奶粉封閉,加Ⅰ抗:β-actin、p-MAPKAPK2、MAPKAPK2、p38MAPK、p-c-jun、c-Jun,4 ℃過夜,TBST洗膜3次,加Ⅱ抗孵育1h,ECL法顯色,凝膠成像系統掃描分析結果。
1.2.3 激光共聚焦顯微鏡觀察內質網及溶酶體形態 LM3、A549細胞常規培養,各實驗組給予不同的處理因素后,對照組用二甲基亞砜(DMSO)做相同處理,用內質網紅色熒光探針和DAPI探針標記內質網,得到內質網斷層掃描圖像;溶酶體紅色熒光探針標記溶酶體,得到溶酶體熒光染色圖像,再經激光共聚焦顯微鏡觀察分析。
2 結 果
2.1 Anisomycin對HepG2細胞空泡的消除作用 實驗組應用不同劑量的p38MAPK和JNK的激活劑Anisomycin(0.25、0.5、1.0μg/mL)作用于 HepG2細胞12h,對照組用DMSO做相同處理。光學顯微鏡下觀察比較,可見Anisomycin對HepG2細胞內自發形成的空泡有消除作用,呈劑量-效應關系,見圖1A。應用0.5μg/mL Anisomycin分別作用于HepG2細胞0、3、6、12h,該消除作用呈時間-效應關系,見圖1B。該結果表明Anisomycin能有效消除HepG2細胞空泡,p38MAPK和JNK信號通路可能參與了細胞空泡化的調節。
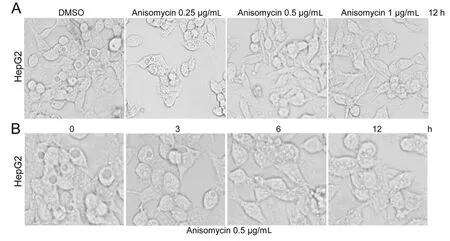
圖1 Anisomycin對HepG2細胞空泡的消除作用(×400)
2.2 Anisomycin通過活化p38MAPK消除細胞空泡 p38 MAPK抑制劑(SB)組和JNK抑制劑(SP)組分別應用5.0 μmol SB203580、20nmol SP600125作用于 HepG2細胞12h,對照組用DMSO做相同處理,Anisomycin組應用0.5μmol/mL Anisomycin作用12h,Anisomycin+SB組和Anisomycin+SP組分別應用5.0μmol SB203580、20nmol SP600125對HepG2細胞預處理1h,再0.5μmol/mL Anisomycin共同作用12h。光學顯微鏡下觀察比較,可見Anisomycin+SB組的明顯空泡化,Anisomycin+SP組與Anisomycin組比較,差異無統計學意義(圖2A)。表明SB203580能阻斷Anisomycin對HepG2細胞空泡的消除作用,SP600125卻不能阻斷該消除作用。Western blot結果顯示,Anisomycin對p38MAPK和JNK的下游效應產物p-MAPKAPK2和p-c-jun的表達水平均有上調作用。這表明了Anisomycin能活化HepG2細胞內p38 MAPK和JNK通路,SB203580選擇性抑制p38MAPK通路,SP600125選擇性抑制JNK通路。結果表明Anisomycin是通過活化p38MAPK消除細胞空泡。
實驗組應用不同劑量的蛋白合成抑制劑CHX(5、10、15 μmol)作用于HepG2細胞12h,對照組用DMSO做相同處理。光學顯微鏡下觀察比較,CHX不能消除HepG2細胞空泡,表明Anisomycin對空泡的消除作用與它的抑制蛋白合成作用無關(圖2B)。
2.3 阻斷p38MAPK誘導LM3細胞空泡形成 應用不同劑量的p38MAPK 選擇性抑制劑 SB203580(0、2.5、5.0、10.0 μmol)作用于LM3細胞12h,誘導空泡形成,呈劑量-效應關系(圖3A)。應用5.0μmol SB203580分別作用于LM3細胞0、6、12、24h,誘導空泡形成,呈時間-效應關系(圖3B)。該結果表明阻斷p38MAPK可誘導LM3細胞空泡形成,呈劑量-時間雙重效應關系。
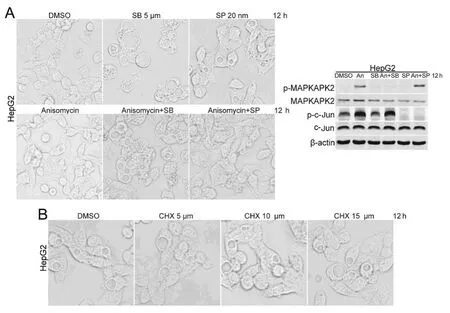
圖2 Anisomycin通過活化p38MAPK消除細胞空泡(×400)

圖3 阻斷p38MAPK誘導細胞空泡形成(×200)
2.4 阻斷p38MAPK誘導多種腫瘤細胞空泡形成 實驗組應用5.0μmol SB203580分別作用于QBC939、LM3、Hela細胞12h,對照組用DMSO做相同處理。光學顯微鏡下觀察比較,可見實驗組細胞內大量空泡形成(圖4)。表明阻斷p38 MAPK可誘導多種腫瘤細胞的空泡形成。
2.5 阻斷p38MAPK介導的空泡形成破壞內質網結構的整體性 實驗組應用5.0μmol SB203580分別作用于LM3、A549細胞12h,對照組用DMSO做相同處理,進行內質網紅色熒光探針和DAPI探針標記內質網,得到內質網斷層掃描圖像。再經激光共聚焦顯微鏡觀察分析,顯示對照組細胞的內質網結構完整,實驗組細胞的內質網結構不完整(圖5)。表明阻斷p38MAPK介導的空泡形成破壞了內質網結構的整體性。
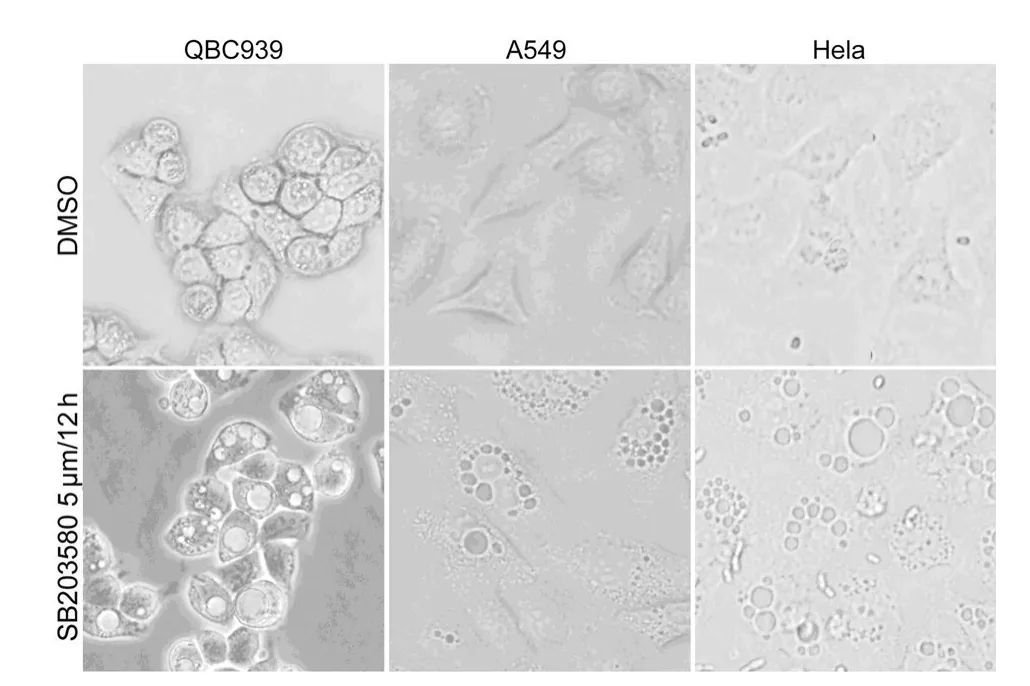
圖4 阻斷p38MAPK誘導多種腫瘤細胞空泡形成(×400)
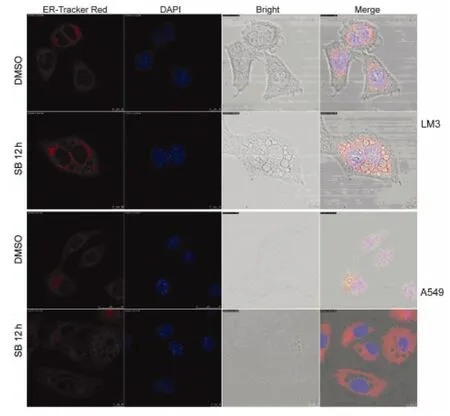
圖5 阻斷p38MAPK介導的空泡形成破壞內質網結構的完整性
2.6 阻斷p38MAPK形成空泡的泡內pH值 應用5.0 μmol SB203580分別作用于LM3、A549細胞12h,進行溶酶體紅色熒光探針標記溶酶體,經激光共聚焦顯微鏡觀察分析,顯示阻斷p38MAPK形成的空泡之中,只有很少一部分呈現熒光染色(圖6)。表明阻斷p38MAPK形成的空泡,只有少數的泡內pH值為酸性。
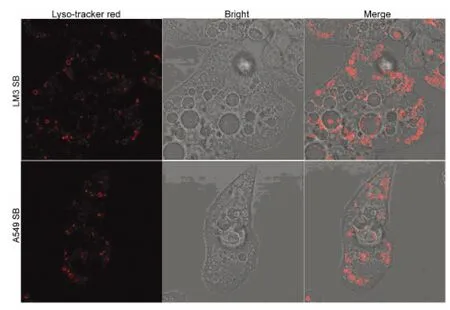
圖6 阻斷p38MAPK形成空泡的泡內pH值
2.7 阻斷p38MAPK介導的空泡形成具有可逆性 實驗組應用5.0μmol SBF203580分別作用于LM3、A549細胞12h,再換不含SB203580的培養基繼續培養6h,對照組用DMSO做相同處理。光學顯微鏡下觀察,可見實驗組經SB203580作用12h,與對照組相比,細胞內有大量空泡形成;而換不含SB203580的培養基6h后,與對照組比較,細胞內空泡化無明顯差異(圖7)。表明阻斷p38MAPK介導的空泡形成具有可逆性。
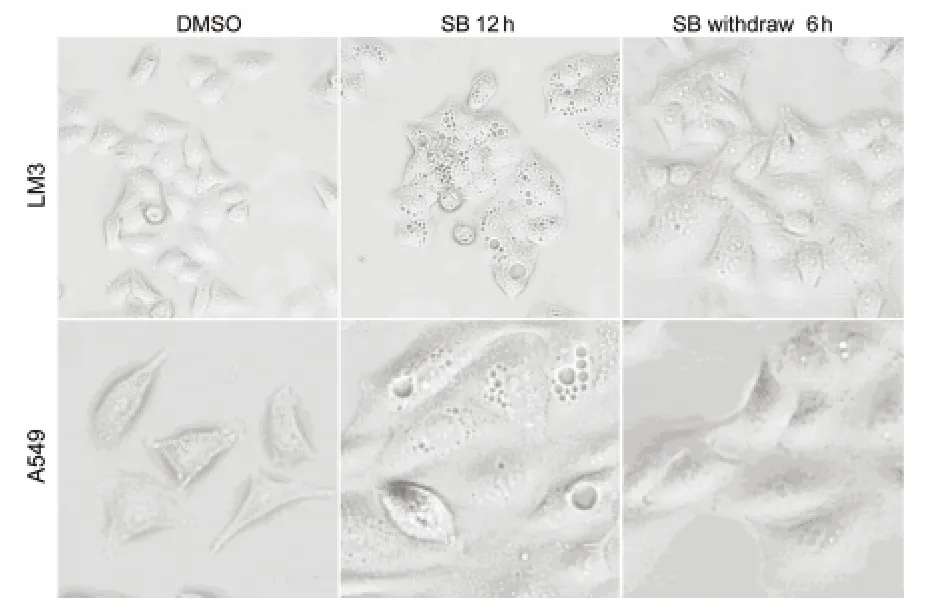
圖7 阻斷p38MAPK介導的空泡形成具有可逆性
3 討 論
細胞空泡化是發生在哺乳類動物細胞中常見的形態學現象之一。細胞局部環境的改變是引起細胞空泡化的根本原因,細胞空泡可自發形成或經誘導刺激而形成。但是,細胞應激信號通路是否參與細胞空泡化,目前尚不明確。本實驗旨在探討p38MAPK通路是否參與了細胞空泡形成。通過研究發現p38MAPK在調節細胞空泡形成中具有重要作用。
應用Anisomycin能有效消除HepG2細胞空泡,因為Anisomycin是p38MAPK和JNK的共同激活劑,這就提示p38 MAPK和JNK信號通路可能參與了細胞空泡化。由于阻斷p38MAPK可抑制Anisomycin對細胞空泡的消除作用,阻斷JNK卻不能抑制Anisomycin的消除作用,提示Anisomycin是通過活化p38MAPK消除細胞空泡的,而不是活化JNK。又因Anisomycin是一種蛋白合成抑制劑,為了確認它對空泡的清除作用是否與抑制蛋白合成有關,本實驗應用了另一種經典的蛋白合成抑制劑CHX作用于HepG2細胞,發現CHX不能消除HepG2細胞空泡,表明Anisomycin對空泡的消除作用與它的抑制蛋白合成作用無關,進一步證明了Anisomycin對空泡的消除作用是通過活化p38MAPK來實現的。
本實驗應用p38MAPK通路的選擇性抑制SB203580阻斷p38MAPK通路,發現可誘導多種腫瘤細胞空泡形成,表明阻斷p38MAPK在細胞空泡形成中發揮了重要作用。最近有研究報道[14]SB202190可以不依賴p38MAPK信號通路介導細胞空泡形成,分析原因可能是因為所用細胞類型和本實驗不同。細胞空泡可在HepG2細胞中自發形成,卻不能自發形成于LM3、A549細胞,可能是不同細胞類型中p38MAPK的活化情況不同。此外,不同細胞類型中的p38MAPK通路的功能也不相同,p38MAPK通路并不是惟一調節細胞空泡形成的通路。因此,p38MAPK通路在細胞空泡化的作用,主要取決于細胞類型,而p38MAPK活化或阻斷的程度,在很大程度上也取決于細胞類型。
本實驗通過激光共聚焦顯微鏡觀察分析內質網斷層掃描圖像,發現阻斷p38MAPK介導的空泡形成破壞了內質網結構的整體性。內質網結構完整性的破壞提示空泡化可能與內質網的應激反應有關。已有報道表明[6-7],細胞空泡就好比是細胞內幫助消化的酸性小區域。本實驗對阻斷p38MAPK形成的空泡進行溶酶體紅色熒光探針標記溶酶體,經激光共聚焦顯微鏡觀察分析,顯示阻斷p38MAPK形成的空泡之中,只有很少一部分呈現熒光染色。表明阻斷p38MAPK形成的空泡,只有少數的泡內pH值為酸性。另外,阻斷p38MAPK可介導空泡形成,當去除阻斷劑細胞空泡則可消除,表明阻斷p38MAPK介導的空泡形成具有可逆性。
本實驗結果提示p38MAPK在調節細胞空泡形成中的重要作用,p38MAPK在調節細胞空泡形成中的分子機制有待于后續實驗進一步探討。
[1]Morissette G,Moreau E,C-Gaudreault R,et al.Massive cell vacuolization induced by organic amines such as procainamide[J].Pharmacol Exp Ther,2004,310(1):395-406.
[2]Wang C,Chen T.Intratumoral injection of taxol in vivo suppresses A549tumor showing cytoplasmic vacuolization[J].Cell Biochem,2012,113(4):1397-1406.
[3]Kar R,Singha PK,Venkatachalam MA,et al.A novel role for MAP1LC3in nonautophagic cytoplasmic vacuolation death of cancer cells[J].Onco Gene,2009,28(28):2556-2568.
[4]Ohkuma S,Poole B.Cytoplasmic vacuolation of mouse peritoneal macrophages and the uptake into lysosomes of weakly basic substances[J].Cell Biol,1981,90(3):656-664.
[5]Henics T,Wheatley DN.Cytoplasmic vacuolation,adaptation and cell death:A view on new perspectives and features[J].Biol Cell,1999,91(7):485-498.
[6]Hiruma H,Kawakami T.Characteristics of weak base-induced vacuoles formed around individual acidic organelles[J].Folia Histochem Cytobiol,2011,49(2):272-279.
[7]Funakoshi T,Aki T,Unuma K,et al.Lysosome vacuolation disrupts the completion of autophagy during norephedrine exposure in SH-SY5Yhuman neuroblastoma cells[J].Brain Res,2012,1490(15):9-22.
[8]Martinet W,De Meyer GR.Autophagy in atherosclerosis[J].Curr Atheroscler Rep,2008,10(3):216-223.
[9]Alonso MM,Jiang H,Gomez-Manzano C,et al.Targeting brain tumor stem cells with oncolytic adenoviruses[J].Methods Mol Biol,2012,797:111-125.
[10]Tasdemir E,Maiuri MC,Tajeddine N,et al.Cell cycle-dependent induction of autophagy,mitophagy and reticulophagy[J].Cell Cycle,2007,6(18):2263-2267.
[11]Zhang Y,Yu C,Huang G,et al.Nano rare-earth oxides induced size-dependent vacuolization:An independent pathway from autophagy[J].Int J Nanomed,2010,5(1):601-609.
[12]Dutta R,Das N.Immunomodulation of serum complement(C3)and macrophages by synthetic pyrethroid fenvalerate[J].In vitro study Toxicology,2011,285(3):126-132.
[13]Wang WB,Feng LX,Yue QX,et al.Paraptosis accompanied by autophagy and apoptosis was induced by celastrol,a natural compound with influence on proteasome,ER stress and Hsp90[J].Cell Physiol,2012,227(5):2196-2206.
[14]Menon MB,Kotlyarov A,Gaestel M.SB202190-induced cell typespecific vacuole formation and defective autophagy do not depend on p38MAP kinase inhibition[J].PLoS One,2011,6(8):e23054.
[15]Takebe K,Nishiyama T,Hayashi S,et al.Regulation of p38MAPK phosphorylation inhibits chondrocyte apoptosis in response to heat stress or mechanical stress[J].Int J Mol Med,2011,27(3):329-335.
