TRPM7參與AngⅡ對心肌細胞鎂離子平衡的調節
2014-09-12 05:40:24郭瑞娜李小青沈之君
東南大學學報(醫學版) 2014年4期
關鍵詞:實驗
郭瑞娜,李小青,沈之君
(南通大學醫學院 病理學與病理生理學系,江蘇 南通 226001)
TRPM7是新近發現的具有離子通道和蛋白激酶雙重結構的雙功能膜蛋白,歸屬于TRPM(transient receptor potential melastatin, 一過性感受器電位通道)超家族。 TRPM7廣泛表達于哺乳動物多種組織器官中,參與體內鎂離子的平衡與代謝。在某些心臟病患者中,TRPM7功能低下可能影響心室肌的傳導和復極化[1]。在房顫患者的心房肌細胞中,TRPM7的功能出現顯著上調[2]。
AngⅡ通過作用于靶細胞膜的AT1(AngⅡ的Ⅰ型)或AT2(AngⅡ的Ⅱ型)受體,參與對血壓及心臟功能的調節。血清AngⅡ長期增高可通過系統和局部作用影響心血管功能,引起高血壓、心肌肥大、心肌炎癥、氧化應激及纖維化等,在心肌重塑和心功能衰竭中起重要作用[3- 4]。
雖然TRPM7和AngⅡ均參與心肌功能的調節,但是兩者之間的關系迄今尚無文獻報道。本研究探討TRPM7是否參與了AngⅡ對心肌細胞鎂離子平衡的調節。
1 材料與方法
1.1 實驗動物
實驗ICR鼠由南通大學實驗動物中心提供。動物實驗中的操作方案及步驟符合南通大學動物實驗中心的規范和要求。動物在密閉的氣室中CO2麻醉后迅速斷頭,開胸取出心臟,用心尖部的心室肌組織作進一步實驗。
1.2 細胞培養
取出生1 d小鼠的心尖部組織,D- Hanks液漂洗3遍,用解剖剪將組織剪碎。剪碎的心肌組織加入0.25%胰酶(Trypsine,Invitrogen),于37 ℃的水浴鍋中孵育10 min,然后加入DMEM培養基終止胰酶的作用,移至離心管中在室溫下離心(1 000 r·min-1,10 min),棄上清,再次加胰酶進行孵育和離心分離,重復2~3次。將分離好的細胞在37 ℃、5%CO2的恒溫培養箱中預培養1.5 h以減少成纖維細胞。取未貼壁的細胞在6孔培養板中進一步培養,培養基為DMEM,細胞密度約為1×106。
1.3 實驗分組及細胞內游離鎂離子動態監測
取分離的小鼠心肌細胞,加入有蓋玻片的6孔培養板中進行原代培養,培養基為DMEM液。培養24 h后用D- Hanks漂洗,在低鎂孵育液中孵育4 h后備用。經低鎂孵育后的細胞分對照組(空白的低鎂孵育液)、AngⅡ組(100 nmol·L-1)、AngⅡ+Losartan組(100 nmol·L-1)及AngⅡ+2- APB組(500 μmol·L-1),上述試劑均購自Sigma- Aldrich公司,按產品說明書配制后直接加入相應的低鎂孵育液中。各組孵育時間均為2 h。孵育結束后按鎂離子熒光探針mag- fura- 2產品說明書(Invitrogen)進行胞內鎂離子標記。從熒光探針標記到實驗結束的時間不超過2 h。
將載有培養細胞的蓋玻片從6孔板中轉移入記錄浴槽,進行細胞內鎂離子動態檢測,記錄浴槽安置在正置顯微鏡的觀察平臺上(Olympus,BX51)并保持持續灌流,滴速為40~50滴·min-1。mag- fura- 2鎂離子熒光探針信號用TTL- 1(Polychrome V,德國)進行雙波激發(波長在340 nm和380 nm之間快速切換),用CCD相機(C10600,日本)實時觀察和記錄510 nm的熒光強度。數據存儲于連線的計算機硬盤中供離線分析。各組細胞先在0.2 mmol·L-1低鎂孵育液灌流下觀察胞內熒光信號,待其穩定后開始記錄。記錄至120 s后換為1.2 mmol·L-1生理鎂孵育液灌流,再記錄至1 800 s后再換成10 mmol·L-1高鎂孵育液灌流,繼續記錄1 800 s后結束實驗。每個細胞的數據在統計時記作n=1。mag- fura- 2熒光探針應用原理是觀察胞內熒光探針在340 nm(熒光探針與鈣離子結合)和380 nm(熒光探針未結合鈣離子)光源激發下其510 nm熒光強度比值(340/380比值)的變化來描述胞內游離鎂離子濃度的相對變化,比值升高提示胞內游離鎂離子上升,比值下降則提示胞內游離鎂離子下降。因此,為了便于組內數據統計和組間數據的比較,我們把每個細胞在0.2 mmol·L-1低鎂孵育液中的340 nm/380 nm比值為基線,分別進行標準化后再作統計分析,實驗結果采用t檢驗。
1.4 實時定量PCR
以出生15 d的小鼠心室肌組織或原代培養心肌細胞為樣本,用Trizol Reagent it(美國Invitrogen公司) 提取樣本總RNA,用TaqMan Probe Assay kit(日本TaKaRa公司) 在Real- time PCR擴增儀(美國ABI)進行實時定量PCR(95 ℃預變性10 min,95 ℃變性15 s,60 ℃退火延伸60 s。共40個循環,每個循環退火末期檢測熒光信號)。小鼠TRPM7 PCR引物:上游5′CTGCCAATCTAGGAGAAGATGCAAT3′;下游引物:5′AGGCGTGTAGTCATTCCTCTTCAAA3′。每組實驗重復3次。實驗結果采用t檢驗。
1.5 Western Blotting檢測
將6孔培養板自培養箱內取出,置于冰上,用PBS漂洗心肌細胞后每孔加入100 μl的蛋白裂解液(內含1 μl的蛋白酶抑制劑)進行處理。然后在4 ℃下進行離心(13 000 r·min-1,25 min),用BCA法測上清中總蛋白濃度,根據每個樣本的總蛋白濃度計算其上樣量,使各樣本間的總蛋白量保持一致。用10%的PAGE膠電泳分離后轉至PVDF膜上,用3%的BSA封閉1 h后加Ⅰ抗Rabbit anti- TRPM7 (以色列Alomone公司),β- actin(美國Abcam公司);4 ℃過夜后用TBST洗膜3次,每次10 min,然后加Ⅱ抗 (Goat anti- rabbit,美國Santa Cruze公司)后在室溫孵育1 h,TBST洗膜3次,每次10 min,加ECL,用凝膠成像系統顯影。每組實驗重復3次。凝膠成像使用灰度統計分析,實驗結果采用成組t檢驗。
2 結 果
2.1 AngⅡ對心肌細胞鎂離子平衡的影響
本實驗觀察到,當對照組的0.2 mmol·L-1低鎂孵育液換成1.2 mmol·L-1生理鎂離子孵育液后,胞內mag- fura- 2的熒光強度比值出現了先快速小幅度上升,在到達最大值(1.2 mmol·L-1Peak)后轉為緩慢下降,在1.2 mmol·L-1低鎂孵育液結束前(1.2 mmol·L-1End)基本回到最初的水平。在10 mmol·L-1高鎂孵育液中,這種緩慢下降的趨勢逐漸消失,在10 mmol·L-1高鎂孵育液結束前(10 mmol·L-1End)比0.2 mmol·L-1低鎂孵育液的比值略低(圖1A),各組mag- fura- 2的熒光強度比值平均值見圖1B,經統計,這些變化差異無統計學意義(圖1C)。
AngⅡ組mag- fura- 2熒光強度比值在1.2 mmol·L-1及10 mmol·L-1中均呈下降的趨勢(圖1A)。經統計,這些變化與對照組比較差異有統計學意義,并且Ang Ⅱ組內0.2 mmol·L-1End、1.2 mmol·L-1Peak、1.2 mmol·L-1End、10 mmol·L-1End間比較差異也有統計學意義(圖1B、C)。AngⅡ誘導的mag- fura- 2熒光強度比值變化可以分別被Losartan或2- APB抑制(圖1A、B)。AngⅡ+Losartan組和AngⅡ+2- APB組的mag- fura- 2熒光強度比值在不同的細胞外鎂離子濃度條件下也出現了有統計學意義的下降(圖1C)。
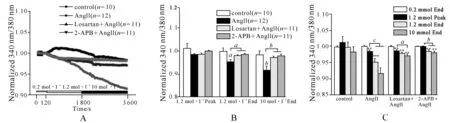
aP<0.05,bP<0.01,cP<0.001
A.各實驗組的細胞內mag- fura- 2的熒光強度比值變化實時動態監測平均值;B.各組mag- fura- 2的熒光強度比值平均值;C.各組mag- fura- 2的熒光強度比值平均值
圖1各組動態熒光強度比值
2.2 AngⅡ對心肌細胞TRPM7表達水平的影響
我們觀察到AngⅡ誘導了心肌細胞游離鎂離子下降,并且這種下降可以被TRPM7通道阻斷劑2- APB抑制。這就說明了TRPM7可能參與了Ang Ⅱ介導的鎂離子外流。因此,我們進一步檢測了Ang Ⅱ對TRPM7表達量的影響。通過qPCR和Western Blotting方法,我們發現Ang Ⅱ沒有影響原代培養心肌細胞中TRPM7的表達(圖2)。
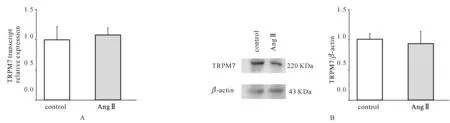
A.小鼠原代心肌細胞TRPM7 mRNA的統計圖(n=3);B.小鼠原代心肌細胞TRPM7蛋白的電泳圖及統計圖(n=3)
圖2AngⅡ對心肌細胞TRPM7表達的影響
3 討 論
我們應用鎂離子熒光探針技術,首次證明了AngⅡ在原代培養心肌細胞中誘導了胞內游離鎂離子下降,而阻斷TRPM7通道則可以顯著抑制這種變化。進一步的分子生物學實驗表明,AngⅡ對心肌細胞中TRPM7的表達量沒有影響。因此,我們的研究提示,AngⅡ可能增強了心肌TRPM7通道對鎂離子的轉運功能。有文獻報道,短時間(5 min)以AngⅡ孵育野生型大鼠的血管平滑肌細胞可以導致胞內Mg2+濃度顯著降低,長時間孵育(24 h)則可以觀察到TRPM7依賴的Mg2+內流,胞內游離Mg2+濃度顯著增加。這種TRPM7依賴性的Mg2+內流可能與AngⅡ誘導的胞內Mg2+濃度持續下降所致的TRPM7反饋性表達增加有關[5]。而Ang Ⅱ 孵育自發性高血壓大鼠血管平滑肌細胞可誘導TRPM7表達下降,進而出現使胞內游離Mg2+顯著降低[6]。此外,我們的實驗數據還顯示,阻斷TRPM7通道并不能完全抑制AngⅡ誘導的鎂離子下降,提示可能有其他鎂離子轉運體的參與的可能性。
通過實驗我們還觀察到,使用AT1受體阻斷劑并不能完全抑制Ang Ⅱ誘導的心肌細胞鎂離子下降。因此,AT1可能參與了AngⅡ對心肌細胞鎂離子的調節,但也不能排除AT2受體參與AngⅡ 對心肌細胞鎂離子調節的可能性。據文獻記載,AT1具有激活心肌細胞增殖、促進心肌纖維化和誘發心肌肥大等作用[7], AT2受體主要介導心肌肥大時炎癥細胞因子表達[8]。但是,AT1受體與TRPM7通道的關系在我們目前的實驗數據中尚不明確。雖然目前已知TRPM7蛋白有多個磷酸化位點,而且TRPM7可以通過自身磷酸化來調節其功能[9]。但是,目前文獻中尚無AT1受體介導的TRPM7蛋白磷酸化的報道。
本實驗證明,AngⅡ可以通過心肌細胞的TRPM7通道下調心肌細胞內游離鎂離子水平。而臨床資料提示,鎂離子不足可能使心臟在高血壓、炎癥和缺血等誘因下更容易受損。我們的實驗為臨床上應用AngⅡ受體阻斷劑治療高血壓、心肌肥大、心律失常等疾病提供了新的思路。
[1] SAH R,MESIRCA P,MASON X,et al.Timing of myocardial trpm7 deletion during cardiogenesis variably disrupts adult ventricular function,conduction,and repolarization[J].Circulation,2013,128(2):101- 114.
[2] SHI J,ZHANG L,ZHANG Y W,et al.Downregulation of doxorubicin- induced myocardial apoptosis accompanies postnatal heart maturation[J].Am J Physiol Heart Circ Physiol, 2012,302(8):H1603- 1613.
[3] BUNKENBURG B,van AMELSVOORT T,ROGG H,et al.Receptor- mediated effects of angiotensin Ⅱ on growth of vascular smooth muscle cells from spontaneously hypertensive rats[J].Hypertension,1992,20(6):746- 754.
[4] NAKAGAMI H,TAKEMOTO M,LIAO J K.NADPH oxidase- derived superoxide anion mediates angiotensin Ⅱ- induced cardiac hypertrophy[J].J Mol Cell Cardiol,2003,35(7):851- 859.
[5] YOGI A,CALLERA G E,ANTUNES T T,et al.Transient receptor potential melastatin 7 (TRPM7) cation channels,magnesium and the vascular system in hypertension[J].Circ J,2011,75(2):237- 245.
[6] TOUYZ R M,HE Y,MONTEZANO A C,et al.Differential regulation of transient receptor potential melastatin 6 and 7 cation channels by ANG Ⅱ in vascular smooth muscle cells from spontaneously hypertensive rats[J].Am J Physiol Regul Integr Comp Physiol,2006,290(1):R73- 78.
[7] AKAZAWA H,KOMURO I.Angiotensin Ⅱ type 1 and type 2 receptor[J].Nihon Rinsho,2009,67(4):687- 694.
[8] ZHOU J,XU X,LIU J J,et al.AngiotensinⅡtype 2 receptors participate in the regulation of inflammatory cytokine secretion in adult rat hypertrophied cardiomyocytes[J].Nan Fang Yi Ke Da Xue Xue Bao,2008,28(11):1971- 1973.
[9] TAKEZAWA R,SCHMITZ C,DEMEUSE P,et al.Receptor- mediated regulation of the TRPM7 channel through its endogenous protein kinase domain[J].Proc Natl Acad Sci USA,2004,101(16):6009- 6014.
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
