凋亡相關因子和腫瘤壞死因子-α介導的細胞凋亡及白介素-19、白介素-10在高原慢性肺心病發病中的作用
2014-08-30 06:19:06楊生岳戴勝歸馮恩志董紅梅劉睿年
中華肺部疾病雜志(電子版) 2014年3期
關鍵詞:血清
楊生岳 戴勝歸 馮恩志 董紅梅 殷 和 張 瑛 賀 巍 劉睿年
高原是一個低氧、寒冷、干燥、日溫差大、紫外線強的特殊環境。在此環境中慢性肺心病(chronic cor pulmonale, CCP)發病率較高,它是嚴重危害高原人群健康的重要慢性呼吸系統疾病。近年研究發現,許多細胞因子、炎性介質和免疫系統參與其發病[1-3]。凋亡相關因子 (factor associated suicide, Fas/Apo-1) 和它的配體FasL是腫瘤壞死因子(tumor necrosis factor, TNF)受體和配體家族中的跨膜糖蛋白[4]。Fas/FasL系統是介導細胞凋亡的主要途徑,凋亡失衡是當今威脅人類健康的許多重大疾病的發病機制之一,其中Fas/FasL基因與肺部疾病關系較為密切[5]。白介素-19(interleukin-19, IL-19)是一種促炎癥細胞因子,能促進中性粒細胞趨化和誘導其它促炎癥細胞因子和趨化因子的釋放[6-7]。而白介素-10(interleukin-10, IL-10)是一種抗炎癥細胞因子,對炎癥反應起著重要的調控作用[8]。本研究的目的是通過探討Fas/ Apo-1和TNF-α介導的細胞凋亡及IL-19、IL-10在高原地區慢性阻塞性肺疾病急性加重期(chronic obstructive pulmonary disease in acute exacerbation, AECOPD)合并CCP發病機制中的作用及與病情程度的關系,為其干預治療提供依據。
資料與方法
一、臨床資料
本研究采用前瞻性隨機、對照研究,所有受試對象均知情同意,并經醫院倫理委員會批準。選擇2013年2月至2013年11月我院住院的高原(海拔2260~3500 m)地區AECOPD合并CCP患者(急性加重組)60例,男性38例,女性22例,年齡54~75歲,平均(59.7±8.2)歲。COPD診斷符合中華醫學會呼吸病學分會制定的《慢性阻塞性肺疾病診治指南(2007年修訂版)》標準,CCP按我國慢性肺心病的標準診斷[9]。經病史、體檢、心電圖、彩色多普勒超聲心動圖、X線胸片、肺功能檢查,排除其他心肺疾病。入院后給予抗感染、祛痰、支氣管擴張劑、氧療等常規綜合治療。其中住院期間死亡3例,其余57例經治療病情緩解后3周為穩定組。健康對照組30例,男18例,女12例,年齡53~72歲,平均(58.8±7.8)歲,為我院門診健康查體者。3組的年齡、性別無統計學差異,均P>0.05。分別測定急性加重組、穩定組和健康對照組患者動脈血氣、肺功能及血清Fas/Apo-1、TNF-α、IL-19及IL-10濃度。
二、研究方法
1. 動脈血氣分析:在未吸氧條件下,取股動脈血2 ml,用血氣分析儀(Opticla-Ⅱ型)測定動脈血氧分壓(partial pressure of artery blood O2,PaO2)、動脈血二氧化碳分壓(partial pressure of artery blood CO2, PaCO2)。
2. 肺功能測定: 用肺功能儀(spirolab-Ⅱ型)測1 s用力呼氣容積(forced expiratory volume in 1 second, FEV1)占預計值的百分比(FEV1%pred),連續測3次,取其最大值計算。
3. 血清Fas/Apo-1、TNF-α、IL-19、IL-10測定:空腹取靜脈血8 ml,置于含10%二乙胺四乙酸二鈉(EDTA-Na2)30 μl、抑肽酶30 μl的抗凝試管中,以離心半徑15 cm,3000 r/min,離心10 min,4 ℃室溫下分離血清,置于-30 ℃凍存備測。血清Fas/Apo-1、TNF-α、IL-19、IL-10測定采用雙抗體夾心ABC-ELISA法,試劑盒由上海森雄科技有限公司提供。所有測定嚴格按說明書操作。
三、統計學分析

結 果
一、急性加重組和穩定組血清Fas/Apo-1、TNF-α、IL-19及IL-10濃度與健康對照組的比較
急性加重組血清Fas/Apo-1、TNF-α、IL-19濃度顯著高于對照組,IL-10濃度顯著低于穩定組和健康對照組(均P<0.01),穩定組與健康對照組比較差異亦非常顯著(均P<0.01),見表1。
二、 急性加重組和穩定組肺功能及動脈血氣與健康對照組比較
急性加重組FEV1%pred、PaO2顯著低于對照組,PaCO2顯著高于穩定組和健康對照組(均P<0.01);穩定組與健康對照組比較差異亦非常顯著(均P<0.01),見表2。
三、急性加重組血清Fas/Apo-1、TNF-α、IL-19、IL-10與肺功能和血氣指標的相關關系
血清Fas/Apo-1、TNF-α、IL-19與FEV1%pred、PaO2顯著負相關(分別r=-0.525、-0.498、-0.603和-0.536、-0.528、-0.463,均P<0.01),與PaCO2顯著正相關(分別r=0.533、0.562、0.498,均P<0.01);IL-10與FEV1%pred、PaO2顯著正相關(分別r=0.476、0.563,均P<0.01),與PaCO2顯著負相關(r=-0.525,P<0.01)。
討 論
在高原低氧環境下,人體心肺功能本身就會發生一系列改變,如肺動脈高壓和心臟優勢的改變;肺通氣、換氣功能的改變;肺泡氣氧分壓(partial pressure of oxygen in pulmonary alveoli, PAO2)、PaO2、PaCO2隨海拔升高而逐漸下降和pH逐漸升高的改變;繼發性紅細胞增多導致血液黏度增高、微循環血流緩慢的改變等等。這些改變隨著海拔的升高和大氣氧分壓的下降而更加明顯。因此,高原肺心病的發病機制、臨床表現、治療及預后等與平原肺心病不完全相同。
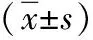
表1 急性加重組和穩定組血清Fas/Apo-1、TNF-α、IL-19及IL-10濃度與健康組比較
表2 急性加重組和穩定組肺功能及動脈血氣與健康組比較
Fas是1989年由Trauth發現的跨膜受體蛋白,1993年正式命名為Apo-1(CD95)抗原[4]。Fas和它的配體FasL是TNF受體和配體家族中的跨膜糖蛋白。Fas與相應的抗體或自然配體FasL結合后能快速誘導細胞凋亡。正常水平的Fas/FasL系統在機體免疫系統中發揮清除不需要的淋巴細胞;限制已發生特異性免疫反應的淋巴細胞克隆;清除那些逃避了胸腺選擇的自身反應性T淋巴細胞等作用。Fas/FasL是介導細胞凋亡的主要途徑,細胞凋亡是細胞程序性死亡過程。凋亡失衡是威脅人類健康的許多疾病的發病機制之一,其中Fas/FasL基因與肺部疾病關系較為密切。Fas/FasL系統除轉導細胞凋亡信號外,在體內還參與促炎癥反應和血管增殖, 如多種炎癥介質的分泌( IL-1、 IL-6、 IL-8)[4]。Xerri等[10]研究發現,正常人肺、食管上皮細胞組織中均有Fas/FasL的表達,進一步研究發現,其主要表達存在于支氣管上皮細胞、肺泡上皮細胞、血管內皮細胞及間質細胞。有研究表明,轉化生長因子-β(transforming growth factor,TGF-β)促進細胞凋亡的作用是通過上調Fas/FasL 的基因表達實現的[11]。IL-19是分布于上皮細胞、內皮細胞、巨噬細胞中的促炎癥細胞因子,主要由活化的單核-巨噬細胞分泌,它能促進中性粒細胞趨化和誘導其它促炎癥細胞因子和趨化因子的釋放[6-7]。而IL-10是一種抗炎癥細胞因子,對炎癥反應起著重要的調控作用,是抑制炎癥過程的重要物質[8]。
本研究結果顯示,高原COPD合并CCP急性加重組患者血清Fas/Apo-1、TNF-α、IL-19水平顯著高于穩定組,穩定組顯著高于健康對照組;急性加重組血清IL-10水平顯著低于穩定組,穩定組顯著低于健康對照組。表明高原COPD合并CCP患者全血和肺內炎癥細胞凋亡異常,促炎癥細胞因子表達增強,而抑炎癥細胞因子表達減弱,這種表現在急性加重期較穩定期更加明顯。其機制可能與缺氧、炎癥、氧化應激等各種有害因素引起肺組織細胞Fas/FasL基因及蛋白表達上調,啟動肺組織細胞死亡程序,從而誘導高原COPD合并CCP患者肺組織細胞凋亡有關。在急性加重期因缺氧、炎癥、氧化應激等有害因素加重,所以表達尤為突出。另外,Fas/FasL系統在誘導支氣管上皮細胞凋亡的同時,也可能誘導促炎癥細胞因子IL-19的分泌和釋放,IL-19作為炎癥介質加重了氣道炎癥反應和肺損傷[7]。這與文獻報道平原COPD患者發生的的結果相一致。Bames等[12]報道,平原COPD重癥患者血漿Fas/FasL顯著增加,其可導致血液和肺內炎癥細胞凋亡異常。劉艷紅等[13]報道,平原COPD急性加重期患者血清IL-6和Fas水平顯著高于健康人。本研究結果還顯示,高原COPD合并CCP患者急性加重期血清Fas/Apo-1、TNF-α、IL-19與FEV1%pred、PaO2呈顯著負相關,與PaCO2呈顯著正相關;IL-10與FEV1%pred、PaO2呈顯著正相關,與PaCO2呈顯著負相關。表明高原COPD合并CCP急性加重期肺功能受損和病情程度與肺內炎癥細胞凋亡異常、促炎癥細胞因子表達增強及抑炎癥細胞因子表達降低相關。同時也表明,高原COPD合并CCP患者體內Fas/FasL倡導的自身反應性T淋巴細胞凋亡不能正常進行,可能也參與了其發病過程。因此,通過Fas/FasL途徑治療高原COPD合并CCP時,不僅要調節肺內炎癥細胞凋亡異常,同時要調節促炎癥細胞因子和抑炎癥細胞因子表達異常。
高原COPD合并CCP患者Fas/FasL基因及蛋白表達上調介導細胞凋亡異常,Fas/Apo-1、IL-19和IL-10可能共同參與了氣道慢性炎癥反應。其肺功能受損和病情程度與肺內炎癥細胞凋亡異常、促炎癥細胞因子表達增強及抑炎癥細胞因子表達降低有關。
參 考 文 獻
1 楊生岳, 郭振援, 馮恩志, 等. 高原地區慢性肺源性心臟病患者肺血管活性因子水平變化及其與肺動脈高壓的關系[J/CD]. 中華肺部疾病雜志: 電子版, 2011, 4(3): 211-215.
2 楊生岳, 馮恩志, 閆自強, 等. 高原慢性阻塞性肺疾病合并慢性肺心病低氧血癥與腫瘤壞死因子系統活性及對營養不良的影響[J/CD]. 中華肺部疾病雜志: 電子版, 2012, 5(3):216-221.
3 楊生岳, 馮恩志, 閆自強, 等. 高原地區慢性阻塞性肺疾病急性加重期合并慢性肺心病抗氧化治療的研究[J/CD]. 中華肺部疾病雜志: 電子版, 2013, 6(1):30-34.
4 Chen JJ, Sun Y, Nabel GJ. Regulation of the proinflammatory effects of Fas ligand (CD95L)[J]. Science, 1998, 282(5394): 1714-1717.
5 楊平滿, 王悅虹, 周建英. Fas/FasL與支氣管哮喘[J]. 國際呼吸雜志, 2011, 31(19): 1475-1478.
6 Chen H, Wang Y, Bai C, et al. Alterations of plasma inflammatory biomarkers in the healthy and chronic obstructive pulmonary disease patients with or without acute exacerbation[J]. J Proteomics, 2012, 75(10): 2835-2843.
7 Hsing CH, Chiu CJ, Chang LY, et al. IL-19 involved in the pathogenesis of endotoxic shock[J]. Shock, 2008, 29 (1): 7-15.
8 Asadullah K, Sterry W, Volk HD. Interleukin-10 therapy-review of a new approach[J]. Pharmaccol Rev, 2003, 55(2): 241-269.
9 中華醫學會呼吸病學分會慢性阻塞性肺疾病學組. COPD診治指南(2007年修訂版)[J]. 中華結核和呼吸雜志, 2007, 30(1): 8-17.
10 Xerri L, Devilard E, Hassoun J, et al. Fas ligand is not only exp ressed in immune privileged human organs but is also coexp ressed with Fas in various ep ithelial tissues[J]. Mol Pathol, 1997, 50(2): 87-91.
11 Kulkarni AB, Karlsson S. Inflammation and TGF beta 1: lessons from the TGF beta 1 null mouse[J]. Res immunol, 1997, 148(7): 453-456.
12 Bames PG, Chowdhury B, Kharitonov SA. et al. Pulmonary biomarkers in chronic obstructive pulmonary disease[J]. Am J Respir Crit Care Med, 2006, 174 (1): 6-14.
13 劉艷紅, 夏熙鄭. IL-6、Fas與慢性阻塞性肺疾病急性加重期的相關性研究[J]. 中國實用醫學, 2009, 4(2):19-20.
猜你喜歡
中老年保健(2021年3期)2021-08-22 06:50:04
天津醫科大學學報(2021年2期)2021-03-29 05:31:08
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
昆明醫科大學學報(2020年12期)2021-01-26 00:44:04
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
豬業科學(2018年8期)2018-09-28 01:27:38
中成藥(2017年8期)2017-11-22 03:18:47
川北醫學院學報(2015年5期)2015-12-05 08:22:29
