HPLC法同時測定復方制劑中烏頭類生物堿含量
2014-08-29 08:14:08王一涵于洋許文迪董雪蓮
江西中醫藥 2014年3期
★ 王一涵 于洋 許文迪 董雪蓮
(長春中醫藥大學 吉林 長春 130117)
該復方制劑為臨床經驗方,是由紅參、附子、甘草等組成的復方中藥制劑,具有益氣養血、通陽復脈之功效,主要用于治療陽氣血弱之心悸病[1]。該制劑現有的質量標準只以甘草中甘草苷為質量評價指標,控制其質量。但其組方中附子即為君藥又為毒性藥物,有必要對其毒性成份進行限定,以確保其用藥安全。本文旨在建立高效液相色譜法同時測定復方制劑中烏頭類生物堿成份的含量測定方法,完善其質量控制指標,為臨床安全用藥奠定基礎。
1 儀器與試藥
1.1 儀器 安捷倫1260高效液相色譜儀,EL204電子天平(萬分之一),XS205電子天平(十萬分之一),GKC-11-CR2電子恒溫水浴鍋(500W)。
1.2 試藥 本實驗所用標準品均來自于中國藥品生物制品檢定所:苯甲酰新烏頭原堿對照品(111795-200901);苯甲酰烏頭原堿對照品(111794-200901);苯甲酰次烏頭原堿對照品(111796-200901);烏頭堿對照品(110720-201111);新烏頭堿對照品(110799-200505);次烏頭堿對照品(110798-200404);甲醇、乙腈、四氫呋喃為色譜純;乙醚、甲醇、鹽酸等為分析純。
2 方法與結果
2.1 色譜條件 色譜柱為Agilent Tc-C18(250mm×4.6mm,5μm),流動相A:乙腈-四氫呋喃(25∶15);流動相B:0.1mol/L乙酸銨溶液(每1 000mL加0.7mL冰醋酸)[2-5]。洗脫程序:0~48min,15%→26%A;48~49min,26%→35%A;49~58min,35%A;58~65min,35%→15%A;65~95min,15%A。柱溫25℃,流速0.8mL/min,檢測波長235nm。
2.2 混合對照品溶液配制 精密稱取苯甲酰新烏頭原堿對照品、苯甲酰烏頭原堿對照品、苯甲酰次烏頭原堿對照品、烏頭堿對照品、新烏頭堿對照品、次烏頭堿對照品4.98,4.96,3.99,4.92,5.08,4.96mg,加0.05%鹽酸-甲醇溶液(V/V)定容于50mL容量瓶內,分別配制成質量濃度為0.0993,0.0978,0.0794,0.0972,0.1016,0.0992mg/mL的對照品母液。分別精密移取上述母液各1mL置于容量瓶內混合均勻,0.22μm微孔濾膜過濾,即得混合對照品溶液。
2.3 供試品溶液制備 取本品粉末,過60目篩,精密稱取3g,置150mL錐形瓶內,加4.5mL氨試液潤濕30min。精密加入100mL乙醚,稱定重量,水浴40℃加熱回流15min,立即冷卻,加乙醚補足失重,密封放置12h,過濾,殘渣加乙醚洗滌兩次,每次10mL,合并乙醚液置于蒸發皿內50℃低溫蒸干,殘渣加0.05%鹽酸-甲醇溶液(V/V)溶解,定容至5mL容量瓶內,用0.22μm微孔濾膜過濾,即得。
2.4 方法學考察
2.4.1 線性關系考察 精密吸取混合對照品溶液2,5,8,10,15,20,25μL,注入液相色譜儀中,以進樣量(μg)為橫坐標,峰面積(A)為縱坐標進行線性回歸,得六混標線性回歸方程如下表1。
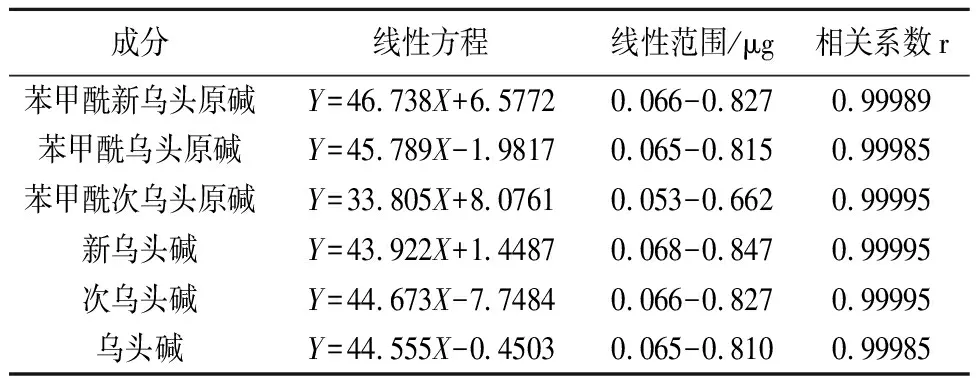
表1 回歸方程和線性范圍
2.4.2 精密度考察 精密吸取混合對照品溶液10μL,照2.1項下色譜條件測定,連續6次,記錄峰面積,計算苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿、新烏頭堿、次烏頭堿、烏頭堿的RSD值分別為:2.8053%,1.7093%,2.7583%,2.3620%,1.6608%,2.6190%,結果表明精密度良好。
2.4.3 穩定性考察 取同一混標溶液分別于0,3,6,12,24,36h按上述液相色譜條件進行測定,得峰面積計算RSD值分別為:2.33%,2.35%,2.66%,1.15%,2.12%,1.44%,結果表明在36h內穩定性良好。取同一供試品溶液同上方法測定,計算苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿、次烏頭堿RSD值分別為:1.61%,1.25%,1.98%,3.02%,表明穩定性良好。
2.4.4 重現性考察 取樣品粉末6份,按2.3項下供試品方法處理,精密吸取10μL,注入液相色譜儀,記錄峰面積,計算含量。苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿、次烏頭堿平均含量分別為:2.1366,0.3903,0.7561,0.0358mg/g,RSD值分別為1.04%,1.59%,0.90%,2.49%,結果表明重現性良好。
2.4.5 加樣回收率考察 取已知含量樣品粉末6份,精密稱定約0.25g,分別精密加入一定量的苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿、次烏頭堿,依法制備供試品溶液,按2.1項下色譜條件測定,計算加樣回收率,結果苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿、次烏頭堿平均加樣回收率分別為:102.14%(RSD2.90%),100.86%(RSD2.73%),101.49%(RSD1.02%),101.71%(RSD2.39%)。
2.4.6 樣品含量測定 取三批不同樣品,按供試品溶液方法制備,按2.1項下色譜條件測定,結果該顆粒中苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿、次烏頭堿含量見表2。
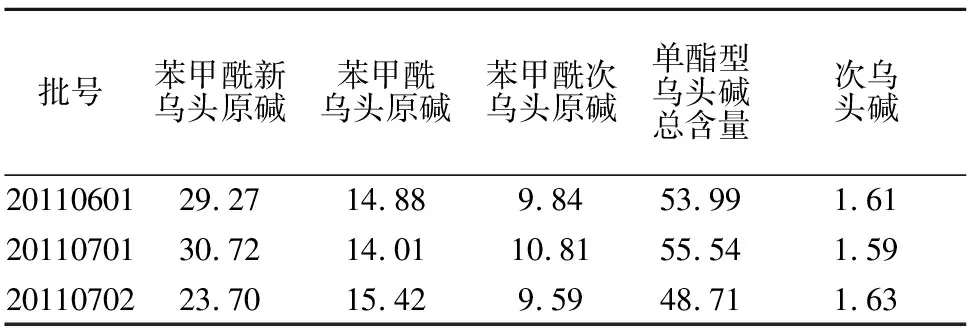
表2 三批復方制劑中烏頭類生物堿含量 /μg·g-1
3 討論
目前對于附子生物堿類成份含量測定方法的文獻報道并不少見。孫蘭[6]等采用乙腈-堿性乙酸銨緩沖溶液為流動相達到了分離效果,但分離時間在65min以上較長,同時堿性(pH=10)流動相不適用于普通色譜柱。陳東安[7]等雖然大大縮短了分離時間,但其分離效果并不十分理想,同時其采用C18固相萃取小柱進行供試品處理過于繁瑣。本實驗所建立的烏頭類生物堿的含量測定方法具有良好的線性關系,科學穩定,高效靈敏,對附子相關制劑質量控制具有一定意義。
本文在藥典方法的基礎上,對流速、流動相等色譜條件稍加改動,達到了良好的分離效果。對于提取方法的優化本文做了大量實驗對比,最終確定乙醚為提取溶劑。采用低溫水浴加熱回流法,較之前的浸泡法大大提高了效率,同時避免了超聲法隨時間水溫增加不易控制的弊病,節約了時間,增加了提取效率。采用0.05%鹽酸甲醇為溶劑,減少了對色譜峰的干擾,分離度較好。本實驗所采用的梯度洗脫程序科學合理,使色譜峰之間都能達到良好分離,同時分離時間適中,50min內即可完成分離,且避免了梯度洗脫普遍易出現的基線漂移現象。
附子由于經過長時間水煎煮等工藝環節后,強毒性的新烏頭堿、烏頭堿受熱水解成單酯型烏頭堿而檢測不出。強毒性的次烏頭堿大部分也都水解了,成品中次烏頭堿含量僅僅為1.6μg/g。強毒性生物堿的總含量遠遠低于藥典規定50多倍,表明該復方制劑臨床應用安全可靠。測得復方制劑中三種單酯型生物堿總含量均高于藥典規定最低限值,表明該復方制劑療效可靠,其含量平均值為52.74μg/g,下浮20%,最終確定復方顆粒三種無毒單酯型生物堿含量下限為不少于40μg/g。本文對復方制劑中附子成份做了含量限定,對其質量評價指標進行補充,為今后該復方制劑控制奠定了基礎。
[1]孫佳欣.強心復脈顆粒制備工藝及質量標準研究[D].長春:長春中醫藥大學,2012:8-46.
[2]國家藥典委員會.中國藥典.一部[S].北京:中國醫藥科技出版社,2010:177.
[3]Zhi-Hong Jiang,Ying Xie,Hua Zhou.Quantification of Aconitum Alkaloids in Aconite roots by a modified RP-HPLC method phytochem icalanalysis[J].Phytochem.Anal,2005(16):415-421.
[4]Liu Hui,Su Juan,Yang Xi,et al.A novel approach to characterize chemical consistency of Traditional ChineseMedicine Fuzi Lizhong pills by GC-MS and RRLC-Q-TOFMS[J].Chinese J Nat Med,2011,9(4):267.
[5]邵峰,俞瑜,任剛,等.HPLC同時測定附子理中丸中3種單酯型生物堿含量[J].中國實驗方劑學雜志,2012,18(16):57-60.
[6]孫蘭,周海燕,趙潤懷,等.HPLC法同時測定附子中6種單酯和雙酯型生物堿[J].中草藥,2009,40(1):131-134.
[7]陳東安,易進海,黃志芳,等.附子煎煮過程中酯型生物堿含量的動態變化[J].中國實驗方劑學雜志,2011,17(3):64-68.
