(PPh3)2Pd(Ar)Br在Suzuki偶聯反應中的應用*
2014-06-23 16:22:14付宏偉李慧李靜戰宏梅程延祥
合成化學 2014年3期
付宏偉,李慧,李靜,戰宏梅,程延祥
(1.中國科學院長春應用化學研究所高分子物理與化學國家重點實驗室,吉林長春 130022;2.中國科學院大學,北京 100039)
(PPh3)2Pd(Ar)Br在Suzuki偶聯反應中的應用*
付宏偉1,2,李慧1,李靜1,2,戰宏梅1,程延祥1
(1.中國科學院長春應用化學研究所高分子物理與化學國家重點實驗室,吉林長春 130022;2.中國科學院大學,北京 100039)
芳基鈀(Ⅱ)化合物(PPh3)2Pd(Ar)Br(1a~1e)與苯硼酸(2)經Suzuki偶聯反應定量合成了一系列取代聯苯(3a~3e),其結構經1H NMR確證。考察了輔助膦配體、反應溫度、反應溶劑、反應時間、原料配比及芳環上取代基對偶聯反應的影響。實驗結果表明,在最佳反應條件[1 1 mmol,n(1)/n(2)=1/1,不加膦配體,甲苯/水為溶劑,于60℃~90℃反應3 h~24 h]下,可定量生成3;過量輔助膦配體和低反應溫度不利于偶聯反應中金屬轉移和還原消除的順利進行,收率較低;芳環上帶有吸電子基團時則有利于金屬轉移和還原消除,收率較高。
芳基鈀(Ⅱ)化合物;Suzuki偶聯反應;金屬轉移;還原消除;電子效應;合成
鈀催化鹵代芳烴與芳基硼酸的Suzuki偶聯反應是構建C-C鍵的重要手段[1-3],已成為合成多烯烴、苯乙烯和聯苯衍生物的有效方法之一,廣泛應用于有機功能材料、醫藥、染料、農藥以及有機合成中間體等領域[4-6]。Suzuki偶聯反應過程主要包含三個基元反應[7],即氧化加成、金屬轉移和還原消除反應。其中,氧化加成是反應歷程的第一步,通常也是決速步驟[8-9],對這一過程的研究相對系統和較為全面[10-12]。而相應的金屬轉移和還原消除過程由于反應速率較快,不利于實驗工作的實施,因而各種反應因素對它們的影響報道較少。
芳基鈀(Ⅱ)化合物(PPh3)2Pd(Ar)Br是Suzuki偶聯反應中由鹵代芳烴(Ar-X)與鈀(0)配合物經氧化加成形成的中間體,分離出這些中間產物并以其為底物與芳基硼酸反應,可以闡明Suzuki偶聯反應中金屬轉移和還原消除過程及其影響因素,為偶聯反應的實際應用提供優化的實驗條件。
本課題組[13-15]利用氧化加成反應合成了一系列芳基鈀(Ⅱ)化合物(1a~1e)。本文以1a~1e為底物與苯硼酸(2)經Suzuki偶聯反應合成了一系列取代聯苯(3a~3e,Scheme 1),其結構經1H NMR確證。并考察了輔助膦配體、反應溫度、反應溶劑、反應時間、原料配比及芳環上取代基對偶聯反應的影響。
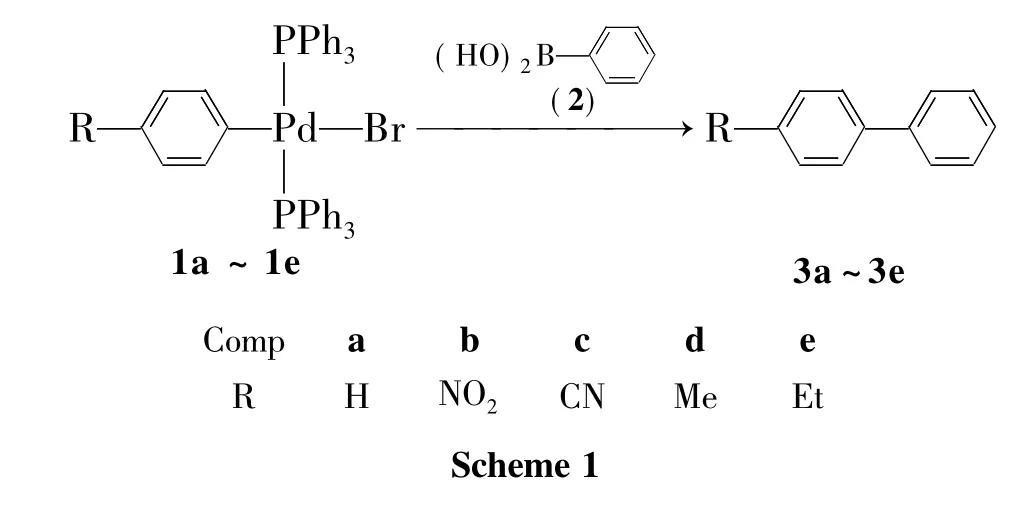
1 實驗部分
1.1 儀器與試劑
Bruker AV 300/400型核磁共振儀(CDCl3為溶劑,TMS為內標)。
1 a~1e按文獻[13-15]方法制備;其余所用試劑均為分析純,其中二氯甲烷(DCM),甲苯和THF均按標準方法除水。
1.2 3的合成通法
在裝有冷凝管的100 mL Schlenk瓶中依次加入1a~1e 1 mol,苯硼酸1 mol,Aliquat 336兩滴,2 mol·L-1K2CO3溶液5 mL和甲苯15 mL,攪拌下于30℃~90℃反應1 h~24 h。冷卻至室溫,分液,有機層用水(3×10 mL)洗滌,無水Na2SO4干燥,旋蒸除溶后經硅膠柱層析[洗脫劑:V(石油醚)∶V(二氯甲烷)=20∶1]純化得白色粉末3a~3e。
聯苯(3a):1H NMR δ:7.53(d,J=7.17 Hz,2H),7.39~7.34(m,2H),7.29~7.25(m,1H)。
4-硝基聯苯(3b):1H NMR δ:8.29(d,J= 8.00 Hz,2H),7.73(d,J=7.92 Hz,2H),7.62 (d,J=7.60 Hz,2H),7.51~7.42(m,3H)。
4-氰基聯苯(3c):1H NMR δ:7.72(d,J= 7.88 Hz,2H),7.57(d,J=8.28 Hz,2H),7.67 (d,J=7.72 Hz,2H),7.50~7.46(m,2H),7.43~7.40(m,1H)。
4-甲基聯苯(3d):1H NMR δ:7.57(d,J= 7.36 Hz,2H),7.48(d,J=7.44 Hz,2H),7.43~7.39(m,2H),7.35~7.29(m,1H),7.24(d,J=7.64 Hz,2H),2.39(s,3H)。
4-乙基聯苯(3e):1H NMR δ:7.57(d,J= 7.74 Hz,2H),7.51(d,J=8.07 Hz,2H),7.41~7.35(m,2H),7.33~7.27(m,1H),7.23(d,J=8.07 Hz,2H),2.66(q,J=7.59 Hz,2H),1.25(t,J=7.56 Hz,3H)。
2 結果與討論
2.1 反應條件優化
以(PPh3)2Pd(Ph)Br(1a)與2的偶聯反應為模板,考察了附加膦配體、反應溫度、反應時間、反應溶劑及原料配比對產物收率的影響,實驗結果見表1。
(1)添加膦配體
在鈀(Ⅱ)化合物催化的Suzuki偶聯反應中,特別是輔助配體為三苯基膦(PPh3)時,通常要加入過量的膦配體,以抑制氧化加成過程中芳基-芳基交換副反應的發生[13]。
1 a 1 mmol,r1=Pd(Ⅱ)/PPh3,r2=n(1a)/n (2)=1/1,甲苯為溶劑,K2CO3為堿,Aliquat 336為相轉移催化劑,于30℃反應24 h,考察添加PPh3對3a收率的影響(No.1~No.4)。從表1可見,反應體系添加PPh3時,3a收率明顯降低。未添加PPh3時,3a收率為81.4%(No.1);隨著PPh3量的增加,收率逐漸降低;當r1=1/3時,收率降至34.9%(No.4),說明過量PPh3不利于金屬轉移和還原消除過程。這與附加膦配體對氧化加成過程的影響恰好相反,其主要原因是由于過量膦配體在金屬轉移和還原消除過程中會包裹和進攻活性鈀(Ⅱ)中心,增強膦配體與中心鈀(Ⅱ)離子的配位能力,抑制輔助配體解離和阻礙兩個芳基形成順式構型,進而減緩還原消除的快速進行,降低偶聯反應產率[7]。
(2)原料配比(r2)
未添加PPh3,其余反應條件同2.1(1),考察r2對3a收率的影響(No.1,No.5~No.7)。實驗結果表明,隨著2用量的增加,3a收率稍有提高(No. 5~No.7),但2過量增加了產物純化難度,同時也造成原料2的浪費。綜合考慮,r2=1/1時較優。
(3)反應溫度
r2=1/1,其余反應條件同2.1(2),考察反應溫度對3a收率的影響(No.1,No.8~No.12)。實驗結果表明,隨著反應溫度升高,3a收率增加; 30℃時收率81.4%;60℃時收率迅速提高至99.6%;繼續升高溫度,收率變化不大(80℃,99.7%);溫度高于100℃,則收率降低,并伴有鈀黑出現。因此溫度升高能夠加速金屬轉移和還原消除過程,但溫度過高時,使得2自身偶聯和脫硼反應加速[7],導致收率下降。值得注意的是,利用氧化加成制備芳基鈀(Ⅱ)化合物時,在低溫(如30℃)反應,無產物生成。
本文偶聯反應的最佳溫度為60℃~90℃。
(4)反應溶劑
反應溫度為60℃或75℃,其余反應條件同2.1(3),分別考察了甲苯/水和THF/水為溶劑時對3a收率的影響(No.9~No.10,No.13~No. 14)。實驗結果表明,溶劑對鈀催化Suzuki反應的影響主要表現在金屬轉移步驟[16]。一般來講,單獨使用極性質子溶劑(如乙醇)或者極性非質子溶劑(如N,N-二甲基乙酰胺)為反應介質時,由于堿在反應體系中的濃度較低,導致金屬轉移過程很難進行。因此,這類反應通常采用有機溶劑與水兩相混合體系,并使用相轉移催化劑Aliquat 336,以加速金屬轉移過程。
從表1可見,以THF代替甲苯為溶劑時,收率變化較小(No.13~No.14),基本接近定量反應,這是由于相轉移催化劑Aliquat 336的使用,弱化了溶劑極性差異所產生的影響。由于甲苯的沸點高于THF,有利于提高反應溫度,因而選取甲苯/水為反應溶劑。
(5)反應時間
甲苯/水為溶劑,反應溫度為65℃,其余反應條件同2.1(3),考察反應時間對3a收率的影響(No.15~No.19)。實驗結果表明,反應時間為1 h時3a收率即可達94.8%;反應3 h獲得了接近定量的收率,與文獻[2]報道一致,也間接證實了氧化加成是Suzuki偶聯反應速率的決速步驟。
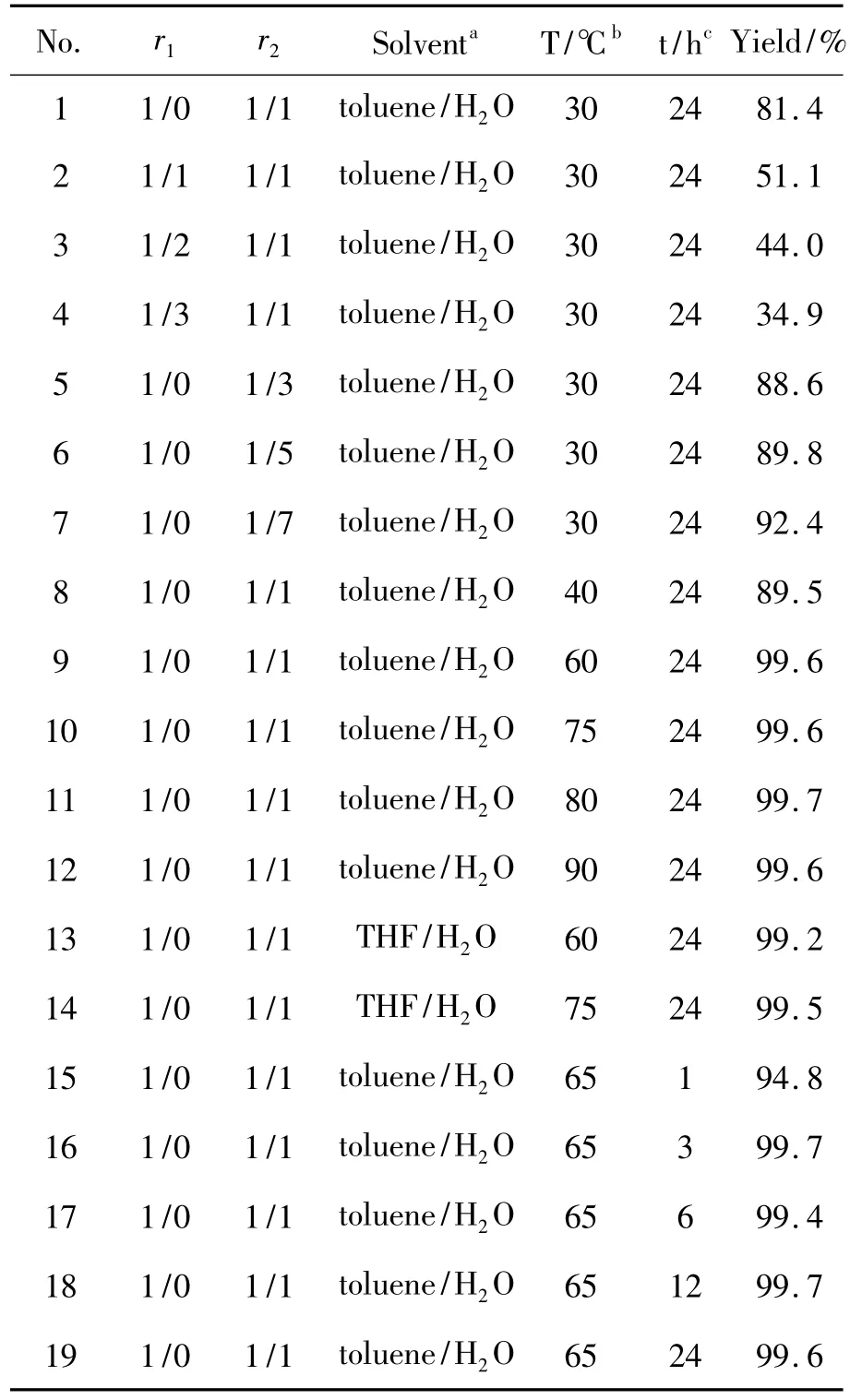
表1 偶聯反應條件優化*Table 1Process optimization of coupling reaction
綜上所述,r2=1/1,不加膦配體,甲苯/水為溶劑,于60℃~90℃反應3 h~24 h,可定量生成偶聯產物3。
2.2 芳環上取代基對偶聯反應的影響
使用芳環上帶有不同取代基的芳基鈀(Ⅱ)化合物(1a~1e)與2反應,對比研究了取代基電子效應對金屬轉移和還原消除過程的影響。在優化的反應條件[1 1 mmol,n(1)/n(2)=1/1,不加膦配體,甲苯/水為溶劑,于60℃~90℃反應3 h~24 h]下反應,盡管取代基不同,但偶聯產物收率均接近定量。在r2=1/1、不附加膦配體和甲苯/水為溶劑,僅縮短反應時間(1 h)和降低反應溫度(30℃)后,偶聯收率明顯降低(7%~18%),且表現出差異,實驗結果見表2。由表2可見,相對于不含取代基的苯基鈀(Ⅱ)化合物參與的偶聯反應,芳環上帶有吸電子基團時有利于反應進行,偶聯產物收率明顯提高,且取代基吸電子能力愈強,收率愈高;而供電子基團則不利于反應進行,偶聯產物收率降低。這與電子效應對氧化加成過程的影響規律相似,也與文獻[16]報道的Suzuki偶聯反應結果一致。吸電子基團之所以能夠提高偶聯產物收率,在于其降低了中心金屬離子的電子云密度,有利于硼酸陰離子的進攻,同時使偶聯產物更易于脫離中心金屬,即加速了金屬轉移和還原消除過程。

表2 芳環上取代基對偶聯反應的影響*Table 2Effects of substituents of aromatic ring on coupling reaction
3 結論
芳基鈀(Ⅱ)化合物(1a~1e)與苯硼酸(2)經直接偶聯反應合成了系列偶聯產物(3a~3e)。并對反應條件進行優化,實驗結果表明,r2=n (1)/n(2)=1/1,不加膦配體,甲苯/水為溶劑,于60℃~90℃反應3 h~24 h,可定量生成偶聯產物;過量輔助膦配體和低反應溫度不利于偶聯反應中金屬轉移和還原消除的順利進行,收率較低;鹵代芳烴上的取代基同樣影響Suzuki偶聯反應進程,吸電子基團有利于偶聯反應中的氧化加成過程[7],因此,研究不同取代基電子效應對反應的影響,可以更全面地理解偶聯反應中的金屬轉移和還原消除過程。研究結果為該類偶聯反應的擴展和應用提供有效的實驗依據。
[1]Nicolaou K,Bulger P G,Sarlah D.Palladium-catalyzed cross-coupling reactions in total synthesis[J]. Angew Chem Int Edit,2005,44:4442-4489.
[2]Suzuki A.Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles,1995-1998[J].J Organomet Chem,1999,576:147-168.
[3]劉望博,胥詩華,范和良,等.兩種新型咔唑衍生物的合成及其光電性能[J].合成化學,2010,18(5): 543-546.
[4]Yamada Y M A,Tekeda K,Takahashi H,et al.Highly active catalyst for the heterogeneous Suzuki-Miyaura reaction:Assembled complex of palladium and non-cross-linked amphiphilic polymer[J].J Org Chem,2003,68:7733-7741.
[5]張揚,吳洋,張燈青,等.新型5-溴嘧啶衍生物的選擇性合成[J].合成化學,2011,19(5):662-664.
[6]吳鋒,楊委,王權,等.鈀催化合成2-(2,4-二氟苯基)吡啶[J].合成化學,2011,19(1):109-110.
[7]Miyaura N,Suzuki A.Palladium-catalyzed cross-coupling reactions of organoboron compounds[J].Chem Rev,1995,95:2457-2483.
[8]Matos K,Soderquist J A.Alkylboranes in the Suzuki-Miyauracoupling:Stereochemicalandmechanistic studies[J].J Org Chem,1998,63:461-470.
[9]Stille J K,Lau K S Y.Mechanisms of oxidative addition of organic halides to group 8 transition-metal complexes[J].Acc Chem Res,1977,10:434-442.
[10]McMullin C L,Jover J,Harvey J N,et al.Accurate modelling of Pd(0)+PhX oxidative addition kinetics [J].Dalton Trans,2010,39:10833-10836.
[11]Senn H M,Ziegler T.Oxidative addition of aryl halides to palladium(0)complexes:A density-functional study including solvation[J].Organometallics,2004,23:2980-2988.
[12]Goossen L J,Koley D,Herman H L,et al.Mechanistic pathways for oxidative addition of aryl halides to palladium(0)complexes:A DFT study[J].Organometallics,2005,24:2398-2410.
[13]Grushin V V.Thermal stability,decomposition paths,and Ph/Ph exchange reactions of[(Ph3P)2Pd(Ph) X](X=I,Br,Cl,F,and HF2)[J].Organometallics,2000,19:1888-1900.
[14]Grushin V V,Alper H.Transformations of chloroarenes,catalyzed by transition-metal complexes[J]. Chem Rev,1994,94:1047-1062.
[15]付宏偉.芳基鈀(Ⅱ)化合物的合成及其在Suzuki縮聚反應中的應用[D].中國科學院長春應用化學研究所,2013.
[16]初文毅,王熳,李新民,等.鈀/碳高效綠色催化Suzuki交叉偶聯反應[J].有機化學,2002,32: 1666-1672.
Applications of(PPh3)2Pd(Ar)Br in Suzuki Coupling Reaction
FU Hong-wei1,2,LI Hui1,LI Jing1,2,ZHAN Hong-mei1,CHENG Yan-xiang1
(1.State Key Laboratory of Polymer Physics and Chemistry,Changchun Institute of Applied Chemistry,Chinese Academy of Sciences,Changchun 130022,China;2.University of Chinese Academy of Sciences,Beijing 100039,China)
A series of substituted diphenyls(3a~3e)were synthesized by Suzuki coupling reaction of (PPh3)2Pd(Ar)Br(1a~1e)with phenylboronic acid(2)under the optimum reaction conditions[1 1 mmol,n(1)/n(2)=1/1,without auxiliary phosphine ligand,using toluene and H2O as a mixture solvent,at 60℃~90℃for 3 h~24 h].The structures were confirmed by1H NMR.The results showed that additional auxiliary phosphine ligand and low reaction temperature were unfavorable to the transmetallation and reductive elimination process,deduced from the lower coupling yield,while the electron withdrawing substituent on the aryl unit could accelerate the aforementioned process,indicative of the higher coupling yield.
aryl Pd(Ⅱ)compound;Suzuki coupling reaction;transmetallation;reductive elimination;electronic effect;synthesis
O625.13;O614.8
A
1005-1511(2014)03-0404-04
2013-03-11;
2014-02-26
國家自然科學基金資助項目(21174141)
付宏偉(1981-),男,漢族,江西德興人,博士研究生,主要從事有機金屬化合物的合成及其催化偶聯反應研究。
戰宏梅,E-mail:hmzhan@ciac.jl.cn
