有機添加劑對堿性條件下電鍍鉍的影響
2013-12-18 05:28:38歐青海戴亞堂張歡
中國有色金屬學報 2013年1期
歐青海,戴亞堂,張歡,馬 麗,馬 歡,申 振
(西南科技大學 四川省非金屬復合與功能材料重點實驗室?省部共建國家重點實驗室培育基地,綿陽 621010)
近年來, 由于半導體鉍擁有一系列的物理和化學性能引起了物理學家和化學家的興趣。鉍薄膜顯示出高效載流、高效載體遷移能力[1]、高磁阻以及量子力學效應[2]和熱電效應[3?7],可以用于量子阱、電流傳感、應用軟件程序材料以及電致變色材料[8?10]。由于其良好的分析性能和“環境友好”的特性,使用薄鉍膜微電極替代水銀?鉛分析,在電化學分析領域很有吸引力的[11?19]。在不久的將來,鉍納米變形特性可以應用于焊接無鉛納米電子器件[20]。
在鉍沉積的研究中,大部分研究者主要集中在超電勢沉積鉍或者鉍成核和生長到非金屬基體上,如碳[15,21]、碲、金、半導體[10,22?23]。同時,電沉積的研究與其他金屬如銅、碲、銀的酸性電解液研究相比,缺少有關有機添加劑的堿性水溶液電沉積鉍的成核行為和生長機制的研究,特別是電沉積鉍早期的電化學行為研究。
本文作者要探究有機添加劑在檸檬酸?乙二胺四乙酸鹽的堿性水溶液對沉積鉍到玻碳電極上的成核和生長機理的影響。電化學技術,如循環伏安法(CV)、計時電流法(CA)可以清晰地了解鉍的成核和生長機理,有助于獲得理想的鉍膜[24];計時電流法(CA)測量的數據廣泛用于Scharifker-Hills方程[25]提取動力學參數。同時, 電鍍鉍到銅基體上可通過掃描電鏡觀察鍍層表面的微觀形貌。
2 實驗
2.1 電沉積試驗
采用有機添加劑的堿性檸檬酸?乙二胺四乙酸的鈉鹽鍍液體系,具體配方為:硝酸鉍80 g/L,乙二胺四乙酸二鈉100 g/L,檸檬酸80 g/L,氨基三亞甲基磷酸2 g/L。當乙二胺四乙酸二鈉濃度過低時,Bi3+在堿性環境中會水解,因此,選擇c(Bi(Ⅲ)):c(EDTA)=1:2,此時,Bi3+可以和乙二胺四乙酸二鈉形成穩定的氨羧酸絡合物[26]。采用不銹鋼片作陽極,經熱處理的光亮紫銅片為陰極。使用SMD?30P(河北大舜)型智能多組換向脈沖電鍍電源進行電鍍,電流密度為 2 A/dm2。電鍍工藝流程為乙醇除油→蒸餾水洗滌→10%稀硫酸除氧化膜→蒸餾水洗滌→磷酸擦拭活化表面→電鍍鉍膜→蒸餾水洗滌→干燥。利用 S4440型(Leica Cambridge LTD)掃描電鏡觀察鍍層的微觀表面形貌。
2.2 電化學測試
本研究采用 AFCP1型電化學工作站(Pine Instrument Company)分別對鍍液體系進行循環伏安及計時安培電流測試,研究鉍沉積機理,探討添加劑對鉍沉積的影響。所有電化學測試都采用三電極體系,以玻碳電極為工作電極,以大面積光亮鉑片為輔助電極,以232型飽和甘汞電極為參比電極,溶液溫度控制在(25±1)℃。
3 結果與討論
3.1 循環伏安
通過循環伏安法試驗可以獲得沉積鉍的電勢區域和沉積特征。圖1(a)所示為有無有機添加劑時電沉積鉍的循環伏安圖。從圖1(a)可以看出,存在有機添加劑時,沉積電位和電流密度明顯增加,可能是有機添加劑的加入使Bi3+形成了更穩定的絡合物,從而對Bi原子沉積的結構、粒度、孔隙度產生影響。圖1(b)所示為存在有機添加劑時不同掃描速度下的循環伏安曲線。從圖1(b)中可以看出,陰極只出現一個波峰,說明 Bi3+沉積是一步三電子還原的過程;同時,還可以看到隨著掃描速度(v)的增加,還原峰的電流密度逐漸增大,還原電流峰的起峰電勢向更負的方向移動,這可能是 Bi3+不能夠滿足快速的動力學成核需求造成的。對于可逆的電極反應過程,還原峰電勢與掃描速度無關[27?28]。因此,圖1的結果表明,電沉積鉍的過程是不可逆的。
對于一個不可逆電化學反應體系,峰電位(Ep)和峰電流密度(jp)滿足下列關系式[29]:
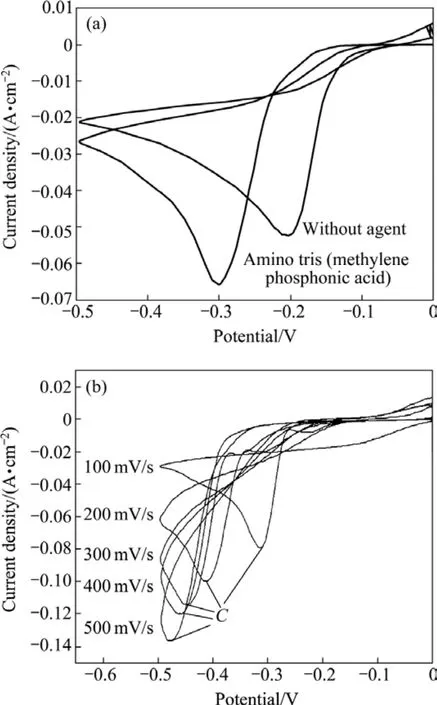
圖1 有無添加劑及有添加劑時不同掃描速率下的循環伏安圖Fig.1 Cyclic voltammograms for bismuth electrodeposition on glassy carbon electrode: (a)v=100 mV/s; (b)Bismuth electrodeposition with organic agent at various scan rates
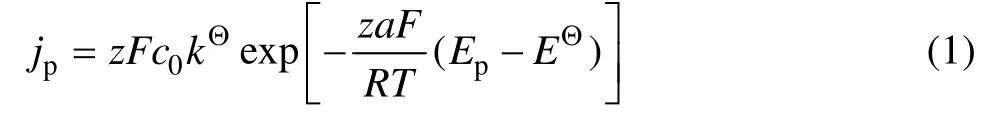
對式(1)取自然對數,變形可得:
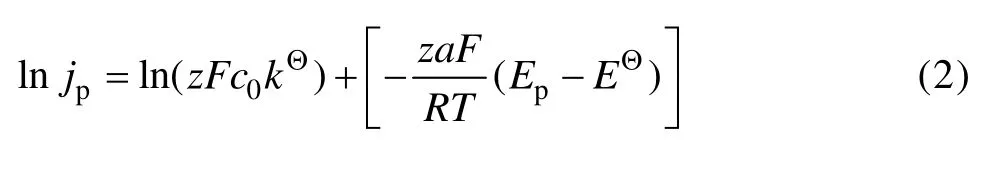
式中:z為為電子轉移數;F為法拉第常數;c0為溶液的本體濃度;kΘ為反應速率常數;α為傳遞系數; R為摩爾氣體常數;EΘ為標準電極電勢;T為熱力學溫度。其中EΘ不隨掃描速率變化而變化,因此,在不同掃描速率下,利用式(2)對ln| jp|—|Ep|作圖(見圖2中的插圖),可以求出傳遞系數α的值約為0.027。
當電極反應的控制步驟為擴散控制時,不同掃描速度下循環伏安曲線的峰值電流密度 (jp)與掃描速度(v)存在以下關系[28]:
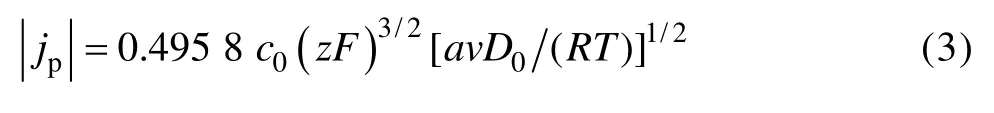
式中:D0為擴散系數。
運用式(3)可以對不同峰值電流密度和掃描速率的平方根(圖1中的C點)作圖,這個峰的峰值電流與相應的掃描速度的平方根的關系如圖2所示。|jp|與v1/2呈良好的線性關系,其相關系數為0.991 4,擬合后成一條直線,可以求出擴散系數為 1.17×10?5cm2/s。因此,可以推斷鉍的電沉積是一個擴散控制的非可逆過程。
3.2 計時電流法
3.2.1 恒電位躍階理論模型
新相形成的一般經歷成核和成長兩個過程,相應的瞬時電流變化可以為電沉積提供有用的動力學信息。在這個過程中,初期階段的成核經歷誘導吸附、原子吸附聚集成簇而形成臨界晶核。電沉積過程成核主要利用Scharifker-Hill(SH)模型進行描述。在電沉積初始階段,當吸附原子進入晶核的過程為速度控制時,Scharifker和 Hill假定在電極上隨機分布的晶核為半球形,且在每個晶核周圍逐漸擴展的擴展區內不能形成新晶核,并考慮到擴展區的重疊、晶核在擴散控制下長大,因此,可以推導出恒電位暫態三維立體成核的公式如下[25,29]:
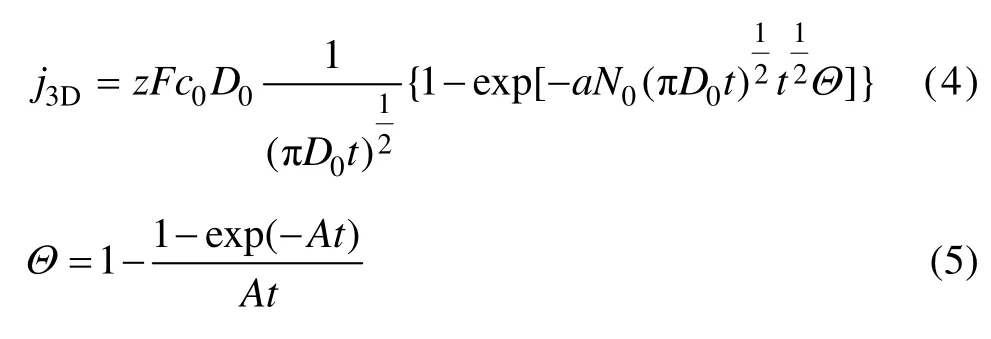
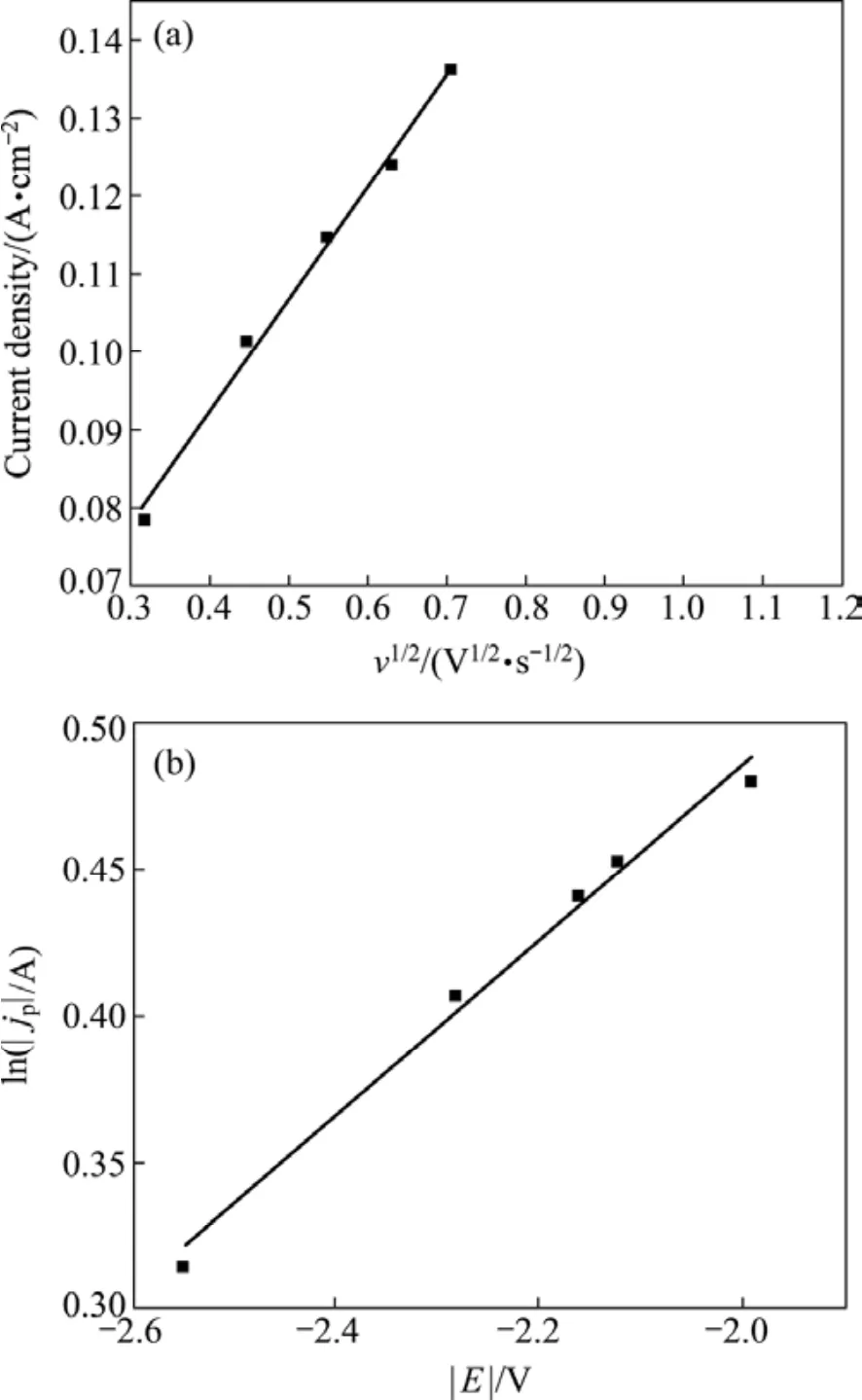
圖2 jp與v1/2的關系圖以及插圖中的ln| jp|—|Ep|曲線Fig.2 Relationship between jp and v1/2 from reductive current peak (a)and ln| jp|—|Ep| curve (b)
式中:j3D為電流密度;N0為最大晶核數密度或表面活性位點數;A為成核速率常數;t為時間。
對于半球形擴散控制條件下的三維多核生長,由式(4)和(5)可以獲得三維連續成核和瞬時成核理論模型來描述暫態下的電流密度增加和減小,為了便于描述這兩種成核方式,將采用極端狀態下的動力學成核,雖然溶液中一些金屬離子的電結晶行為并不能完全適用于這兩種極端情況[5],但在很多情況下該模型具有一定的適用性,如將這兩種方式轉化成無因次函數即(j/jm)2~ t/tm理論成核關系式:
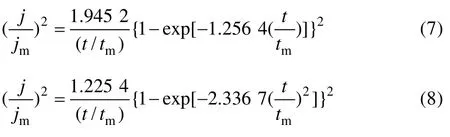
式中:j為電流密度;jm為最大電流密度;tm為最大電流密度所對應的時間。式(7)描述連續成核,式(8)描述瞬時成核。
在恒電位階躍分析中,電位從開路電位開始階躍到金屬的沉積電位,在這樣的條件下,游離金屬離子到電極表面為傳質速度控制,體系從沒有反應到形成穩態,可以利用Cottrell方程描述如下:

在電位階躍的極短時間內,由于雙電層充電導致電流密度先迅速增加隨后減小,而后由于晶核的形成和新相的生長,電流再次逐漸增加并達到最大值,隨后又出現衰減, 此時整個電極表面表現為擴散控制[30?32]。
3.2.2 電沉積鉍的初期行為
不同階躍電位下,堿性有機添加劑鍍液中的鉍電沉積成核和生長的恒電位曲線如圖3所示。由圖3可以看到,當電位低于?500 mV時,由于電極雙電層充電導致與時間暫態曲線中電流密度有所增加;當電位高于?500 mV時,由于晶核的形成和新相的生長,才觀察到電結晶形核/生長引起的電流密度逐漸增加;當電位高于?600 mV時,電流密度先呈指數式增加并達到最大值,然后出現對數式減小,最后達到平衡。隨著階躍電位的負移,即過電位增大,峰電流密度相應的增大,其對應的tm值(出現峰電流所需時間)呈規律性的縮短,說明過電位升高導致成核速率加快,成核誘導時間縮短。
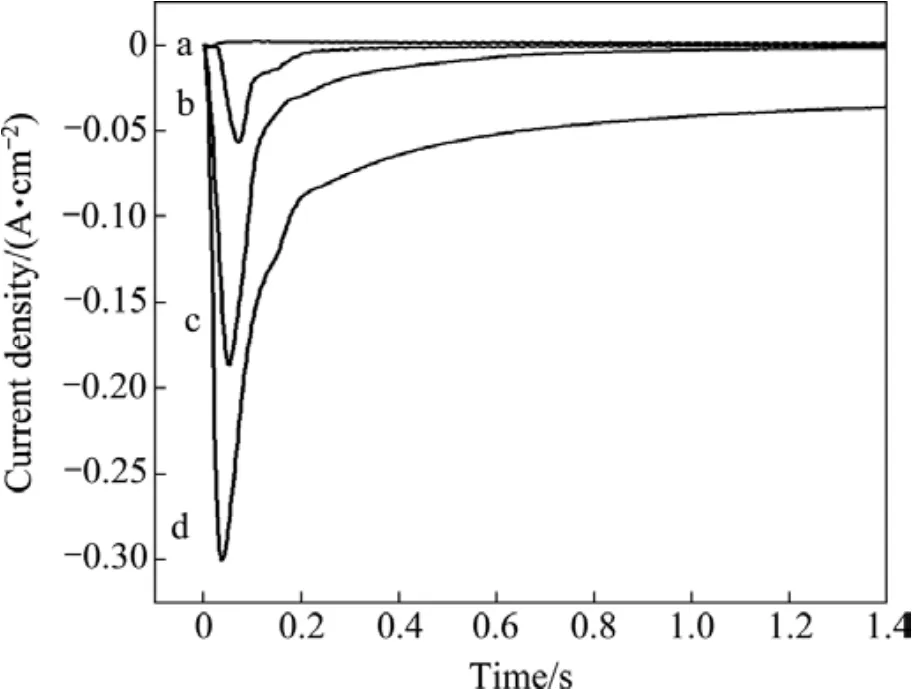
圖3 不同電勢下計時電流圖Fig.3 Current transients for bismuth electrodeposition on glassy carbon electrode stepped from 0 mV to different potentials:(a)?500 mV; (b)?600 mV; (c)?700 mV; (d)?800 mV
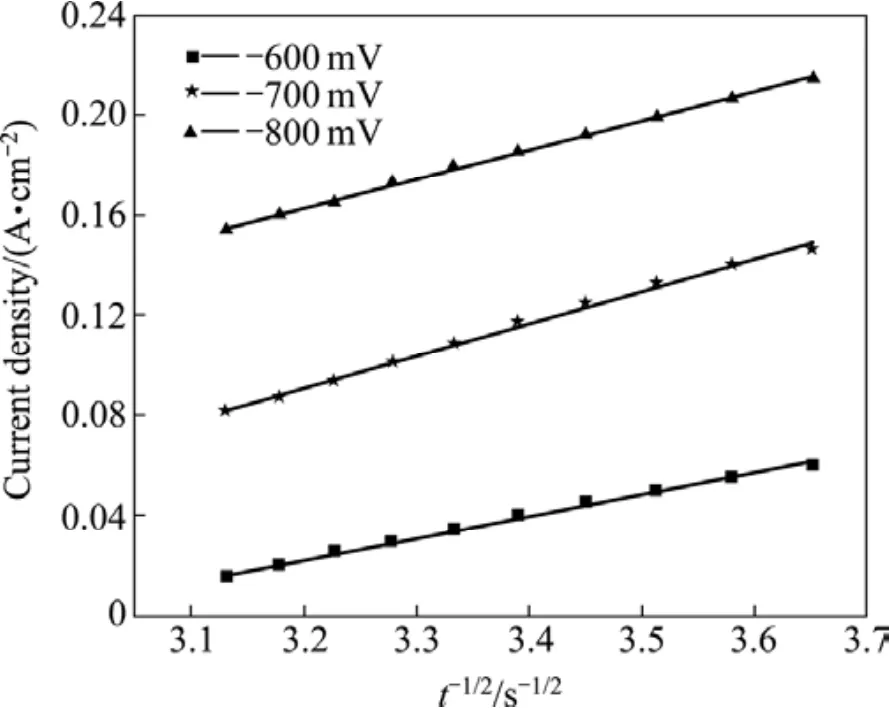
圖4 Cottrell方程相對應的j—t?1/2關系圖Fig.4 Relationship between j and t?1/2 from Cottrell equation
圖4是由圖3暫態曲線的上升部分所對應的j—t?1/2關系圖。由圖4可知,在較高的過電位下,j與 t?1/2呈線性關系,此時整個電極表面反應表現為擴散控制。由等式(9)的 Cottrell方程,可以求得在?600 ~ ?800 mV的擴散系數分別為1.46×10?5、 1.52×10?5和1.67×10?5cm2/s。從所得結果來看,不同電勢對擴散系數的影響較小,本研究得到擴散系數的平均值為1.55×10?5cm2/s。這與用式(3)求得的值(1.17×10?5cm2/s)比較接近。因此,循環伏安法和計時電流法都是適用于有機添加劑沉鉍的動力學研究。
循環伏安曲線可以為計時電流的超電勢沉積作指導,而Scharifker-Hill(SH)模型可以從定性上分析實驗中暫態成核方式。圖5所示為在超電勢下的成核,實線和虛線分別表示理論上的三維連續成核和瞬時成核。圖5(a)所示為在?800 mV超電壓下有機添加劑對成核的影響。由圖5(a)可以看出,鍍液中不論是否存在有機添加劑,鉍的電沉積機理都接近三維連續成核機理。結合循環伏安圖可知,有機添加劑只改變了鉍的沉積電勢,不影響其電沉積機理。圖5(b)所示為在不同超電勢下鉍的成核實驗。從圖5(b)可以看出,隨著電勢的升高,鉍的電沉積機理更加接近三維連續成核,可能是隨著階躍電位的增大,電極表面由于鉍的沉積而產生大量的活性點。這些活性點形成新的晶核,晶核在擴散控制下長大。另外,從圖5(b)還可以看出,當t/tm小于1.2時,實驗曲線與三維理論連續成核重合比較好,超過這段時間,實驗曲線低于理論曲線,可能是體系自身存在輕微擾動[32]。
4 添加劑對鉍鍍層形貌的影響
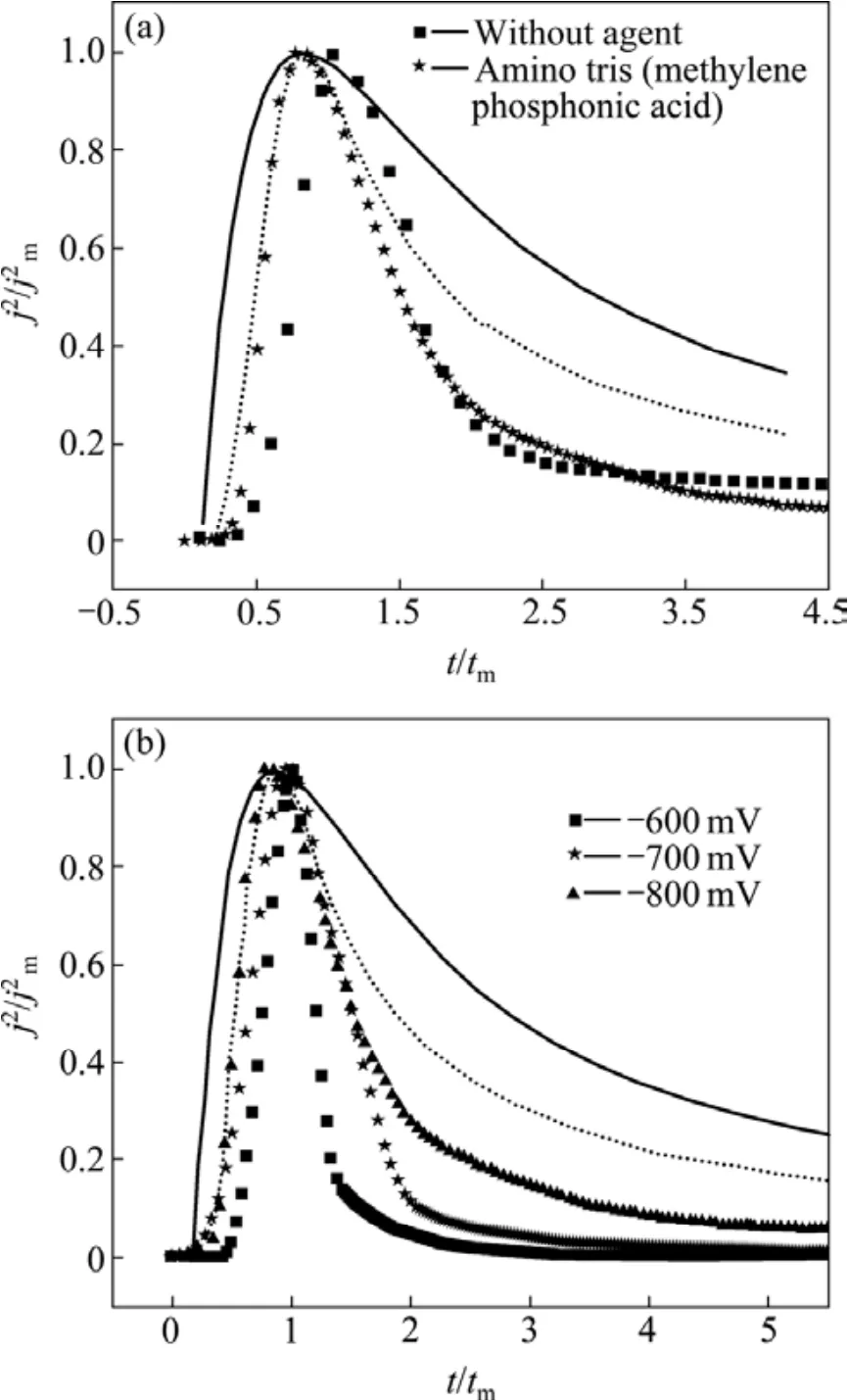
圖5 電沉積鉍過程的無因次j2/j2m—t/tm 曲線Fig.5 Non-dimensional j2/j2m—t/tm curves of data for bismuth electrodeposition on glassy carbon electrode: (a)With potential of ?800mV (vs SCE); (b)Potential range from ?600 mV to ?800 mV (vs SCE).(Theoretical transients for instantaneous (solid line)and progressive (dotted line)nucleation were calculated according to Scharifker–Hills model.)
圖6所示為鍍液中加入有機添加劑前后制備的鉍鍍層的SEM像。如圖6(a)所示,鍍液中加入有機添加劑之前,所制備的鉍鍍層表面凹凸不平,較粗糙,晶粒大小約為10 μm,呈貝殼狀,晶粒間有直徑約5 μm左右的深溝,同時可以看出貝殼狀晶核頂端生長有大量的小晶粒。因此,可認為此體系為連續成核機理,在鉍的電沉積過程中,主要是不斷長出新的晶粒覆蓋在晶核頂端,而在晶粒的生長底部,三維擴散場所覆蓋的電極表面因溶液的貧化和電場屏蔽很難形成新的晶核以至于鍍層凹凸不平。如圖6(b)所示,鍍液中加入有機添加劑之后,由于有機添加劑具有良好的微觀平整效應,所制備的鉍鍍層表面形貌明顯改善,非常平滑,可能是由于氨基三亞甲基膦酸對Bi3+的螯合作用,限制了鍍液中自由金屬離子的濃度,減慢了金屬離子的還原速度,從而細化鍍層晶粒,提高了鍍層表面致密度和光滑度。此時的鉍鍍層在放大5 000倍條件下已基本看不出單個晶粒。可見有機添加劑的加入對鉍鍍層表面形貌的改善十分顯著。

圖6 鉍鍍層的SEM像Fig.6 SEM images of Bi films: (a)Without organic agent; (b)With amino tris(methylene phosphonic acid)
5 結論
1)循環伏安法測試表明,鉍沉積過程主要是受擴散控制的非可逆過程的影響。根據不同掃描速率可以求得Bi3+的擴散系數為1.17×10?5cm2/s。
2)電沉積初期行為分析表明,鉍電結晶遵從擴散控制下的三維 Scharifker-Hill連續成核模型;隨著過電位的增大,成核活性點增多,形核弛豫時間縮短;當電沉積趨于穩定,即階躍電位大于?600 mV時,Bi3+的平均擴散系數約為1.55×10?5cm2/s,與循環伏安法測試出的擴散系數接近,說明這兩種方法可用于有機添加劑沉積鉍的動力學研究。
3)通過電沉積的方法,從堿性有機添加劑的檸檬酸—乙二胺四乙酸鹽鍍液體系中獲得鉍鍍層。SEM觀察表明,氨基三亞甲基膦酸的加入降低了鍍液中自由金屬離子的濃度,對鉍的沉積具有一定的阻礙作用,使沉積電位增加,從而細化了鍍層晶粒,使鍍層表面更加的平整、致密。
[1]JIANG Shan, HUANG Yun-hui, LUO Feng, DU Nan, YAN Chun-hua.Synthesis of bismuth with various morphologies by electrodeposition[J].Inorganic Chemistry Communications,2003, 6(6): 781?785.
[2]CHO S, KIM Y, OLAFSENC L J, VURGAFTMANC I,FREEMAN A J, WONG G K L,.MEYER J R, HOFFMAN C A,KETTERSON J B.Large magnetoresistance in post-annealed polycrystalline and epitaxial Bi thin films[J].Journal of Magnetism and Magnetic Materials, 2002, 239(1/3): 201?203.
[3]ZIEGLER J P.Status of reversible electrodeposition electrochromic devices[J].Solar Energy Materials & Solar Cells,1999, 56(3/4): 477?493.
[4]CóRDOBA DE TORRESI S I, CARLOS I A.Optical characterization of bismuth reversible electrodeposition[J].Journal of Electroanalytical Chemistry, 1996, 414(1): 11?16.
[5]RICHOUX V, DILIBERTO S, BOULANGER C, LECUIRE J M.Pulsed electrodeposition of bismuth telluride films: Influence of pulse parameters over nucleation and morphology[J].Electrochimica Acta, 2007, 52(9): 3053?3060.
[6]LI Liang, ZHANG Yong, LI Guang-hai, ZHANG Li-de.A route to fabricate single crystalline bismuth nanowire arrays with different diameters[J].Chemical Physics Letters, 2003, 378(3/4):244?249.
[7]LI Liang, YANG You-wen, FANG Xiao-sheng, KONG Ming-guang, LI Guang-hai, ZHANG Li-de.Diameter-dependent electrical transport properties of bismuth nanowire arrays[J].Solid State Communications, 2007, 141(9): 492?496.
[8]GOLIA S, ARORA M, SHARMA R K, RASTOGI A C.Electrochemically deposited bismuth telluride thin films[J].Current Applied Physics, 2003, 3(2/3): 195?197.
[9]XIAO Feng, HANGGARTERA C, YOOB B, RHEEMA Y,LEECK K-H, MYUNG N V.Recent progress in electrodeposition of thermoelectric thin films and nanostructures[J]. Electrochimica Acta, 2008, 53(28):8103?8117.
[10]SANDNES E, WILLIAMS M E, BERTOCCI U, VANUDIN M D, STAFFORD G R.Electrodeposition of bismuth from nitric acid electrolyte[J].Electrochimica Acta, 2007, 52 (21): 6221?6228.
[11]ARDUINI F, CALYO J Q, AMINE A, PALLESCHI G,MOSCONE D.Bismuth-modified electrodes for lead detection[J].Trends in Analytical Chemistry, 2010, 29(11):1259?1304.
[12]REHACEK V, HOTOVY I, VOJS M.Bismuth-coated diamond-like carbon microelectrodes for heavy metals determination[J].Sensors and Actuators B, 2007, 127(1):193?197.
[13]HUTTON E A, OGOREVC B, HOCEVAR S B, WELDON F,SMYTH M R, WANG J. An introduction to bismuth electrode for use in cathodic electrochemical detection[J].Electrochimistry Communication, 2001, 3 (12): 707?711.
[14]WANG Joseph, LU Jian-min.Bismuth film electrodes for adsorptive stripping voltammetry of trace nickel [J].Electrochimistry Communication, 2000, 2 (6): 390?393.
[15]KRóLICKA A, R.PAULIUKAIT?, ?VANCANCARAC I,METELKA R, BOBROWSKI A, NORKUS E, KALCHER K,VYTRAS K.Bismuth-film-plated carbon paste electrodes[J].Electrochimistry Communication, 2002, 4(2): 193?196.
[16]HUTTON E A, HOCEVAR S B, OGOREVC B, SMYTH M R.Bismuth film electrode for simultaneous adsorptive stripping analysis of trace cobalt and nickel using constant current chronopotentiometric and voltammetric protocol[J].Electrochimistry Communication, 2003, 5(9): 765?769.
[17]KEFALA G, ECONOMOU A, VOULGAROPOULOS A,SOFONIOU M.A study of bismuth-film electrodes for the detection of trace metals by anodic stripping voltammetry and their application to the determination of Pb and Zn in tapwater and human hair[J].Talanta, 2003, 61(4): 603?610.
[18]KRRóLICKA A, BOBROWSKI A.Bismuth film electrode for adsorptive stripping voltammetry–electrochemical and microscopic study[J].Electrochimistry Communication, 2004,6(2): 99?104.
[19]HUTTON E A, VAN ELTEREN J T, OGOREV B, SMYTH M R.Validation of bismuth film electrode for determination of cobalt and cadmium in soil extracts using ICP-MS[J].Talanta, 2004,63(4): 849?855.
[20]BUREK M J, JIN S, LEUNG M C, JAHED Z, WU J,BUDIMAN A S, TAMURA N, KUNZ M, TSUI T Y.Grain boundary effects on the mechanical properties of bismuth nanostructures[J].Acta Materialia, 2011, 59(11): 4709?4718.
[21]YANG Min-li, HU Zhong-bo.Electrodeposition of bismuth onto glassy carbon electrodes from nitrate solutions[J].Journal of Electroanalytical Chemistry, 2005, 583(1): 46?55.
[22]OSIOVICH N P, STRELTSOY E A, SUSHA A S.Bismuth underpotential deposition on tellurium[J].Electrochimistry Communication, 2000, 2: 822?826.
[23]JEFFREY C A, HARRINGTON D A, MORIN S.In situ scanning tunneling microscopy of bismuth electrodeposition on Au(111)surfaces[J].Surface Science, 2002, 512(1/2):L367?L372.
[24]SADALE S B, PATIL P S.Nucleation and growth of bismuth thin films onto fluorine-doped tin oxide-coated conducting glass substrates from nitrate solutions[J].Solid State Ionics, 2004, 167(3/4): 273?283.
[25]SCHARIFKER B R, HILLS G.Theoretical and experimental studied of multiple fucleation[J].Electrochimica Acta, 1983, 28(7): 879?889.
[26]SOTO A B, ARCE E M, PALOMAR-PARDAVé M,GONZáLEZ I.Electrochemical nucleation of cobalt onto glassy carbon electrode[J].Electrochimica Acta, 1996, 41(16):2647?2655.
[27]DOLATI A, AFSHAR A, GHASEMI H.A kinetic study on the electrodeposition of cadmium with the presence of organic agents in sulfate solutions[J].Materials Chemistry and Physics,2005, 94(1): 23?28.
[28]CHEN Zeng, ZHANG Mi-lin, HAN Wei, HOU Zhi-yao, YAN Yong-de.Electrodeposition of Li and electrochemical formation of Mg-Li alloys from the eutectic LiCl–KCl[J].Journal of Alloys and Compounds, 2008, 464(1/2): 174?178
[29]EMERY S B, HUBBLEY J L, ROY D.Voltammetric and amperometric analyses of electrochemical nucleation:electrodeposition of copper on nickel and tantalum [J].Journal of Electroanalytical Chemistry, 2004, 568: 121?133.
[30]趙旭山, 譚澄宇, 陳文敬, 劉 宇, 李勁風, 鄭子樵.Ni-SiC復合鍍層電結晶初期動力學分析[J].中國有色金屬學報,2008, 18(5): 823?828.ZHAO Xu-shan, TAN Cheng-yu, CHEN Wen-jing, LIU Yu, LI Jin-feng, ZHENG Zi-qiao.Nucleation kinetics analysis of Ni-SiC composite film during early electrocrystallization processes[J].The Chinese Journal of Nonferrous Metals, 2008,18(5): 823?828.
[31]胡 煒, 譚澄宇, 崔 航, 鄭子樵.Sn-Cu合金的電沉積行為及添加劑的影響[J].中國有色金屬學報, 2010, 20(5):1006?1011.HU Wei, TAN Cheng-yu, CUI Hang, ZHENG Zi-qiao,Electrodeposition behavior of Sn-Cu alloy and effect of additives on deposition process[J].The Chinese Journal of Nonferrous Metals, 2010, 20(5): 1006?1011.
[32]GRUBA? Z, METILO?-HUKOVI? M.Electrodeposition of thin sulfide films: nucleation and growth observed for Bi2S3[J].Thin Solid Films, 2002, 413(1/2): 248?256.
